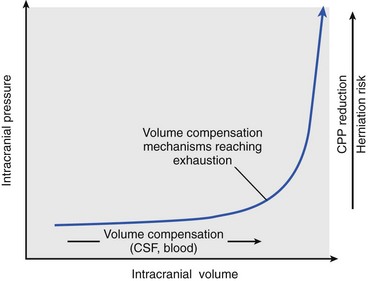
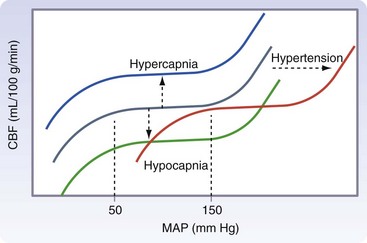
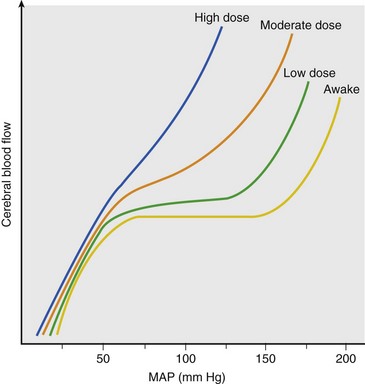
Chapter 38 This compensatory mechanism as described in the Monro-Kellie doctrine is a limited one. Because the cranium is by nature a noncompliant vault, once the compensatory mechanisms have been exhausted, a given increase in the volume of one factor will result in a dramatic increase in intracranial pressure. The relationship between intracranial pressure and volume is shown in Figure 38-1. It should be noted that patients with a space-occupying lesion may be situated more to the right on the compliance curve shown in Figure 38-1 than those without such a lesion. This means that although small increases in intracranial volume may be tolerated in healthy patients, individuals situated toward the right of this curve may decompensate as a result of further increments in intracranial volume. Cerebral perfusion pressure therefore can be reduced by decreases in mean arterial pressure (hypotension) or increases in intracranial pressure (see next section) or central venous pressure (caused by coughing or vomiting). In a study by McPherson et al.39 with two groups of beagle dogs anesthetized with thiopental and maintained on isoflurane, cerebral perfusion pressure was 82 ± 5 mm Hg (10.9 ± 0.7 kPa) (mean ± standard error of the mean [SEM]). A number of studies on human patients with severe head injuries have shown an increase in mortality and poor outcome when cerebral perfusion pressure is less than 60 mm Hg (8 kPa) for a sustained period of time.73 Intracranial pressure (ICP) is the pressure exerted by tissues and fluids within the skull. Under normal circumstances, intracranial pressure ranges from 5 to 12 mm Hg (0.7 to 1.6 kPa).43 In a study by Mazzoni et al.37 with eight mongrel dogs anesthetized with acepromazine and pentobarbital and mechanically ventilated with 70% N2O in oxygen to maintain arterial carbon dioxide tension at approximately 35 mm Hg (4.7 kPa), intracranial pressure was 8 ± 0.5 mm Hg (1.1 ± 0.07 kPa) (mean ± SEM). Intracranial hypertension (elevated intracranial pressure) may result from an expanding tissue or fluid mass, depressed skull fracture, interference with normal absorption of cerebrospinal fluid, or systemic disturbances promoting brain edema. Normal cerebral blood flow in the dog is 75.9 ± 10.4 mL/min/100 g (mean ± SEM).35 Autoregulatory mechanisms maintain a relatively constant cerebral blood flow despite changes in systemic arterial pressure (Figure 38-2). However, this autoregulation fails if systemic mean arterial pressure falls below 60 mm Hg.35,46 From this point on, cerebral blood flow declines linearly with systemic arterial pressure. This implies that these patients are more susceptible to cerebral ischemia resulting from low mean arterial pressure. Similarly, at systemic mean arterial pressures higher than ≈180 mm Hg, cerebral perfusion pressure can augment linearly with arterial pressure.46 In such cases, an abrupt increase in systemic arterial pressure can result in a dangerous elevation of intracranial pressure. It is worth noting that this autoregulation can become impaired or abolished by a variety of insults, including trauma, hypoxemia, hypercapnia (Figure 38-3), and large-dose volatile anesthetics (Figure 38-4). Moreover, some patients with traumatic brain injury have a rightward shifted lower limit of autoregulation, necessitating the maintenance of a higher mean arterial pressure than normal.73 CO2: A decrease in PaCO2 will cause vasoconstriction of cerebral blood vessels. Thus, a reduction in PaCO2 by hyperventilating can cause a rapid decrease in cerebral blood flow and intracranial pressure (see Figure 38-2). In fact, mild hyperventilation (PaCO2 ≈30 mm Hg) is a commonly used technique to decrease cerebral blood flow and intracranial pressure. Additional decreases in cerebral blood flow do not seem to occur when PaCO2 is <25 mm Hg (3.3 kPa).36 Prolonged periods of hypocapnia have been shown to worsen outcome in patients with traumatic brain injury.43 Furthermore, Marion et al.36 stated that even short periods of moderate hyperventilation (as short as 30 minutes) can significantly increase extracellular concentrations of secondary brain injury mediators (i.e., glutamate, pyruvate, and lactate), potentially as a result of reduction in cerebral blood flow. Because of hypoperfusion and accumulation of brain injury mediators, a PaCO2 <30 mm Hg (4 kPa) in compromised brain tissue may lead to additional brain injury.36 In addition, aggressive hypocapnia can shift the oxygen dissociation curve farther to the left, making oxygen less available to the tissues. Cerebral metabolic rate for oxygen (CMRO2) is the volume of O2 metabolized by the brain per unit of time. Published normal values in the dog are on the order of 3.5 ± 0.3 mL/min/100 g.39 When considered with respect to total body oxygen consumption, the brain accounts for approximately 20% of the total body oxygen requirement. Sixty percent of the CMRO2 is used in generating adenosine triphosphate (ATP) to support neuronal activity, and 40% is used for maintaining cellular integrity. Syring et al.64 found correlation in dogs and cats presented with head trauma between blood glucose concentrations on admission and severity of their neurologic condition. However, in this study, the degree of hyperglycemia could not be associated with outcome. Adverse metabolic and cerebral ischemic effects of high blood glucose concentrations are well documented. The influence of hyperglycemic ischemia on tissue damage and cerebral blood flow was studied in rats subjected to short-lasting transient middle cerebral artery occlusion.17 Brief focal cerebral ischemia combined with hyperglycemia led to larger and more severe tissue damage. The mechanistic basis for this remains unclear. The most persistent hypothesis is that glucose, in the absence of oxygen, undergoes anaerobic glycolysis, resulting in intracellular acidosis, which amplifies the severity of other deleterious cascades initiated by the ischemic insult. A great deal of emphasis has been placed on the minor differences in anesthetic-induced changes in cerebral blood flow, cerebral metabolic rate, and intracranial pressure that have been consistently demonstrated in a variety of studies. In a study of almost 700 human patients, the drugs used for induction or maintenance of anesthesia were not independent risk factors for intraoperative brain swelling. The risk factors identified for intraoperative brain swelling included intracranial pressure at the start of surgery, the degree of midline shift on computed tomography scan, and histologic diagnosis of glioblastoma or metastasis.55 The drugs most frequently employed for craniotomy in human patients include local anesthetics, the opioids sufentanil and remifentanil, propofol, and the alpha-2 agonist dexmedetomidine and clonidine.20 However, a number of agents can be safely and successfully used to anesthetize patients for neurosurgery. Dexmedetomidine is a drug that is rapidly gaining popularity in neuroanesthesia. Dexmedetomidine is a highly selective alpha-2-adrenoceptor agonist that has been shown to have both sedative and analgesic effects. Research in laboratory mice has shown that the sedative properties of alpha-2-adrenoceptor agonists are mediated solely via the alpha-2A subtype receptor. Norepinephrine release from the locus coeruleus was inhibited when a hypnotic dose of dexmedetomidine was administered.28 Dexmedetomidine offers a unique “cooperative sedation” (the patient easily transitions from sleep to wakefulness when stimulated, allowing assessment of mental status), anxiolysis, and analgesia with virtually no respiratory depression. Dexmedetomidine has been shown to provide good hemodynamic stability during intracranial tumor surgery, attenuating the response to intubation and emergence.66 Additionally, dexmedetomidine has shown neuroprotective properties (decreased cortical ischemic volume by 40%) in an experimental model of cerebral ischemia in the rat when compared with normal saline.26 A significant improvement in functional neurologic outcome after ischemia-hypoxia–induced brain injury has also been demonstrated by Ma et al.33 in the rat. In this study, dexmedetomidine exhibited dose-dependent protection against brain matter loss and improved neurologic functional deficits (prehensile traction, strength, balance, and coordination) induced by the hypoxic-ischemic insult. Rates of 1 µg/kg/hr were used in dogs by Lin et al.,32 in combination with propofol infusion (0.25 mg/kg/min) or isoflurane (2%), with a discrete increase (yet significant) or no change in mean arterial pressure. Acepromazine: Acepromazine is a phenothiazine neuroleptic and antipsychotic agent. It blocks postsynaptic dopamine receptors in the central nervous system and may inhibit the release and increase the turnover rate of dopamine. It is believed to depress portions of the reticular activating system, which assists in the control of alertness, body temperature, basal metabolic rate, emesis, vasomotor tone, and hormonal balance. Additionally, acepromazine has anticholinergic, antihistaminic, antispasmodic, and alpha-adrenergic blocking effects. The effects of phenothiazines on intracranial pressure in dogs with reduced intracranial compliance have not been evaluated. Tobias et al.67 found no evidence that acepromazine increased the risk of epileptic episodes in dogs with a history of seizures. Ketamine: The phencyclidine derivative ketamine is a noncompetitive N-methyl-D-aspartate (NMDA) receptor antagonist with the thalamo-neocortical projection system as the primary site of action. The effects of ketamine on cerebral blood flow and cerebral metabolic rate are regionally specific; in the limbic structures, cerebral blood flow and cerebral metabolic rate increase, whereas within the cortex, reductions in cerebral blood flow and cerebral metabolic rate occur. Ketamine also increases the release of catecholamines, resulting in elevated cardiac output and arterial blood pressure. Administration of ketamine has been related to increases in cerebral blood flow and intracranial pressure; therefore, it is routinely avoided as an induction agent for neuroanesthesia. Recent literature suggests that a reevaluation of the use of ketamine in patients with increased intracranial pressure may be indicated. The effect of ketamine on intracranial pressure was studied in normocapnic, mechanically ventilated pigs by Schmidt et al.58,59 Racemic ketamine did not impair cerebral blood flow autoregulation when used alone. When used in a model with artificially increased intracranial pressure, racemic ketamine (10 mg/kg) decreased intracranial pressure a maximum of 10.8%. Results from a meta-analysis conducted by Himmelseher and Durieux21 revealed that greater cerebral perfusion pressure is maintained and the need for vasopressors is reduced when ketamine is used for sedation rather than opioids. During anesthesia, ketamine does not impair cerebral autoregulation. However, this may not be true in the presence of N2O.21 Fentanyl: This pure µ-agonist is a widely used agent in neuroanesthesia. Fentanyl, even at high dosages (25 µg/kg IV), did not alter cerebral vascular response to changes in PaO2, PaCO2, and systemic blood pressure in a study by McPherson and Traystman41 in dogs. When the effects of alfentanil, fentanyl, and remifentanil on hemodynamic and respiratory variables in human patients undergoing supratentorial craniotomy were compared, no significant differences, except reduced time to eye opening in the remifentanil group, were found.9 Alfentanil: Alfentanil is a short-acting analog of fentanyl. Even at high dosages (0.32 mg/kg IV), alfentanil did not alter cerebral vascular response to changes in PaO2, PaCO2, and systemic blood pressure in a study by McPherson et al. in dogs.40 Remifentanil: Remifentanil is an ultrashort-acting opioid. It is unique among opioids in that it is hydrolyzed by non-specific plasma and tissue esterases; its pharmacokinetics are independent of hepatic and renal functions. Remifentanil half-life in the dog is approximately 7 minutes.27 Bilotta et al.5 compared two different anesthetic protocols in human beings with supratentorial expanding lesions: propofol-remifentanil and propofol-sufentanil. It was concluded that propofol-remifentanil allowed an earlier cognitive recovery; however, the two groups had similar intraoperative hemodynamics and awakening and extubation times. The effects of a constant infusion of midazolam on cerebral function, metabolism, and hemodynamics were studied in dogs by Fleischer et al.14 The CMRO2 and cerebral blood flow decreased until a plateau was reached at approximately 75% of control values.
Anesthesia for Intracranial Surgery
Physiology
Cerebral Perfusion Pressure
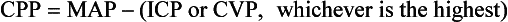
Intracranial Pressure
Cerebral Blood Flow
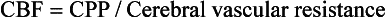
Effects of Chemical Factors on Cerebral Blood Flow
Cerebral Metabolic Rate for Oxygen
Glycemia
Pharmacology in Neuroanesthesia
Intravenous Agents
Dexmedetomidine
Opioids
Benzodiazepines
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Anesthesia for Intracranial Surgery
Only gold members can continue reading. Log In or Register to continue
