Shock
Shock is the clinical picture observed when tissue oxygen delivery or utilization is compromised. Oxygen delivery (DO2) depends upon adequate cardiac output (CO) and arterial oxygen content (CaO2). Oxygen utilization reflects the ability of the cells to gain access to oxygen and convert it into energy. Tissue hypoxia is the result of inadequate oxygen delivery or utilization. The body responds to tissue hypoxia or shock by engaging a constellation of compensatory mechanisms to preserve vital organ function and host viability. These compensatory mechanisms are manifest as the classic clinical findings in a patient in shock: (1) tachycardia (to increase oxygen delivery), (2) tachypnea (to increase oxygenation), (3) peripheral vasoconstriction (to maintain perfusion of vital organs), and (4) mental depression (in response to decreased perfusion or hypoxia). Clinical findings result from cellular, tissue, organ, and systemic sensing and response to tissue hypoxia. Early sensors of shock detect alterations in cardiac output and arterial oxygen content, and activate the initial hemodynamic response. As shock persists, tissue oxygen debt ensues. Cellular oxygen sensing is activated, leading to changes in energy metabolism and gene expression. Failure to restore tissue oxygenation will result in cell death and progression to irreversible shock.
Shock can be classified according to its main contribution to impaired tissue oxygenation (Box 6-1 and Table 6-1). Most forms of shock impair oxygen delivery. Cardiogenic shock is used to describe conditions that impair forward flow of blood from the heart. Although sometimes described as “obstructive shock,” conditions that restrict right ventricular filling may also be grouped under cardiogenic shock. Hypovolemic shock occurs when circulating volume is inadequate. Distributive shock is caused by inappropriate vasodilation, resulting in inadequate effective circulating volume. Septic shock is one form of distributive shock, but its pathophysiology is complex, and it can result in impairment of cardiac function, fluid loss, and impaired oxygen utilization. Hypoxic shock results from inadequate arterial oxygen content or impaired mitochondrial function (impaired oxygen uptake). A list of abbreviations and equations used in this chapter is provided in Tables 6-2 and 6-3.

Table • 6-2
Abbreviations Used in This Chapter
| 2,3-DPG | 2,3-Diphosphoglycerate |
| ABP, mm Hg | Arterial blood pressure |
| ACTH | Adrenocorticotropic hormone |
| ADH | Antidiuretic hormone |
| ADP | Adenosine diphosphate |
| aPTT | Activated partial thromboplastin time |
| ARDS | Acute respiratory distress syndrome |
| ARNT | Aryl hydrocarbon nuclear translocator |
| ATP | Adenosine triphosphate |
| AVP | Vasopressin (also called antidiuretic hormone, or ADH) |
| CaO2, mL/dL | Arterial oxygen content |
| cDO2, mL/min | Critical oxygen delivery |
| CO, L/min | Cardiac output |
| CO2 | Carbon dioxide |
| CRH | Corticotropin-releasing hormone |
| CvO2, mL/dL | Venous oxygen content |
| CVP, mm Hg | Central venous pressure |
| DAP, mm Hg | Diastolic arterial pressure |
| DIC | Disseminated intravascular coagulation |
| DO2, mL/min | Oxygen delivery |
| ECG | Electrocardiogram |
| EDP | End-diastolic pressure |
| EDV | End-diastolic volume |
| FAST | Focused assessment with sonography for trauma |
| FiO2 | Fractional inspired oxygen |
| H+ | Hydrogen ion |
| H2O | Water |
| Hb, g/dL | Hemoglobin |
| HCO3− | Arterial bicarbonate |
| Hct, % | Hematocrit |
| HIF | Hypoxia-inducible factor |
| HMGB | High mobility group box protein |
| HR, bpm | Heart rate |
| HRE | Hypoxia response element |
| ICAM | Intercellular adhesion molecule |
| IL | Interleukin |
| LDH | Lactate dehydrogenase |
| LiDCO | Lithium dilution cardiac output |
| MAP, mm Hg | Mean arterial pressure |
| MODS | Multiple organ dysfunction syndrome |
| NaCl | Sodium chloride |
| NAD+ | Nicotinamide adenine dinucleotide |
| NADH | Coenzyme formed by reaction of NAD+ with oxidizing agent |
| O2 | Oxygen |
| O2ER, % | Oxygen extraction ratio |
| OPS | Orthogonal polarization spectral imaging |
| PAI-1 | Plasminogen activator inhibitor |
| PaO2, mm Hg | Arterial oxygen partial pressure |
| PCO2 | Partial pressure of carbon dioxide |
| PCV | Packed cell volume |
| PiCCO | Transpulmonary thermodilution cardiac ouput |
| PIRO | Predisposition, infection, response, organ failure |
| PO2 | Partial pressure of oxygen |
| PT | Prothrombin time |
| PvO2 | Mixed venous oxygen tension |
| rhaPC | Recombinant human activated protein C |
| RR | Respiratory rate |
| SaO2, % | Arterial hemoglobin oxygen saturation |
| SAP, mm Hg | Systolic arterial pressure |
| ScvO2, % | Central venous oxygen saturation |
| SIRS | Systemic inflammatory response syndrome |
| SpO2, % | Oxygen saturation of hemoglobin in peripheral blood as measured by pulse oximetry |
| StO2 | Muscle tissue oxygen saturation |
| SV, mL | Stroke volume |
| SvO2,% | Hemoglobin saturation of mixed venous blood |
| SVR | Systemic vascular resistance |
| TNF | Tumor necrosis factor |
| tPA | Tissue plasminogen activator |
| VO2, mL/min | Oxygen uptake |
Table • 6-3
| Oxygen delivery | DO2 (mL/min) = CO (L/min) × CaO2 |
| Cardiac output | CO = SV × HR |
| Arterial oxygen content | CaO2 = Hb × SaO2 × 1.34 + PaO2 × 0.003 |
| Venous oxygen content | CvO2 = Hb × SvO2 × 1.34 + PvO2 × 0.003 |
| O2ER | O2ER = VO2/DO2 × 100 |
| MAP | DAP + 1/3 × (SAP − DAP) |
Pathophysiology of Impaired Oxygen Delivery and Oxygen Uptake
Oxygen is the essential fuel for efficient energy production in aerobic metabolism. Unfortunately, tissues lack the ability to store oxygen; therefore a continuous supply of oxygen is required to match metabolic demand. The basic factors determining oxygen delivery are arterial oxygen content and cardiac output.
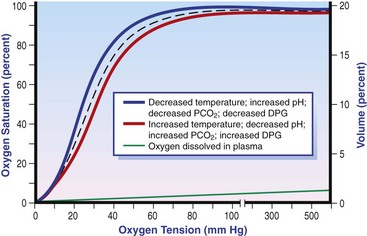
The solubility of oxygen in plasma is low; therefore, the major determinant of arterial oxygen content is hemoglobin. The ability to carry oxygen is influenced by the amount of hemoglobin. The saturation of hemoglobin is dependent on the function of the hemoglobin molecule (i.e., its ability to take up oxygen in the lung and release it into the tissues) and of gas exchange in the lung (Figure 6-1). Cardiac output is a reflection of the ability of the heart to pump blood through the lungs for oxygenation, and then through the remainder of the body for oxygen delivery, and is influenced by heart rate (HR) and stroke volume (SV).
Systemic or global oxygen delivery (DO2) reflects the total volume of oxygen delivered to the tissues per minute, irrespective of how blood flow is distributed in the regional circulation:
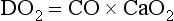
Determinants of Oxygen Delivery
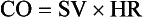
where SV equals volume of blood ejected per single cardiac cycle, and HR equals heart rate in beats/min.
Cardiac output is the volume of blood ejected by the heart into the systemic circulation each minute. Cardiac output is calculated from the product of stroke volume and heart rate. In human patients, cardiac output is measured in L/min. However in veterinary patients, because of the highly variable sizes and shapes of patients, cardiac output is often expressed in terms of mL/kg/min, or as the correlated cardiac index.36 Cardiac index is obtained by dividing the cardiac output by the body surface area and is therefore expressed as mL/m2/min. Because the pulmonary and systemic circulations occur in series, in normal physiology, the cardiac output of the left and right heart is equivalent. Acute increases in cardiac output occur through increases in heart rate. Tachycardia is effective for short-term increases in cardiac output, but it occurs at a cost. Increases in heart rate require shortening of the cardiac cycle. Systole requires a fixed amount of time; therefore diastole is disproportionately reduced during tachycardia. The consequences of shortened diastole include reduced filling time, which can limit stroke volume and reduce cardiac perfusion (the coronaries are perfused during diastole). Most chronic responses to increased cardiac output rely on increases in stroke volume. Factors that determine stroke volume are preload, afterload, and contractility.
Afterload is the force that opposes muscle contraction, and in cardiac muscle, it is equivalent to the ventricular wall tension developed during systole. It can also be described as pressure in the left ventricle required to eject blood into the systemic circulation. Afterload is influenced mainly by systemic vascular resistance. Low blood pressure is the major determinant of decreased afterload. A decrease in blood pressure will reduce the tissue perfusion pressure, as defined by the difference between arterial and venous pressures. Decreased tissue perfusion results in decreased tissue oxygenation. Increasing blood pressure with vasopressor agents will increase perfusing pressures but can have the untoward effect of increasing the work of the heart.
Contractility refers to the force and velocity of cardiac muscle contraction when the conditions of preload and afterload remain constant. The initial neuroendocrine response to shock will increase cardiac contractility. Although contractility is an intrinsic factor of cardiac muscle, afterload and preload are dependent on the myocardium and on vascular status and thus are defined as functional coupling factors between heart and blood vessels.
Blood flow is not directly measured, nor factored into cardiac output. cardiac output, as a global measure, assumes that blood flow is uniform in all tissue beds. Blood flow, however, is influenced by regional vasomotor function, circulating volume, and activation of blood components (e.g., coagulation, platelet aggregation, neutrophil adhesion). Insufficient flow can result in regional or global tissue ischemia, which, in addition to tissue hypoxia, leads to depletion of glucose and other metabolic fuels, as well as accumulation of byproducts of metabolism (e.g., carbon dioxide [CO2], lactate).
Arterial Oxygen Content
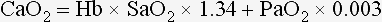
where 1.34 is a constant representing the amount of oxygen (mL) carried by 1 g of fully saturated hemoglobin (Hb), and 0.003 is the oxygen solubility coefficient at 37° C.
Oxygen (O2) content depends mainly on hemoglobin concentration (in g/dL) and oxygen saturation of hemoglobin in arterial blood (SaO2; measured as %). SaO2 is the percentage of available hemoglobin that is bound to oxygen. Oxygen binds hemoglobin in a cooperative relationship (i.e., hemoglobin affinity for oxygen increases as the oxygen saturation of hemoglobin increases). For this reason, the oxygen-hemoglobin dissociation curve, which relates hemoglobin saturation of O2 to the partial pressure of oxygen (PO2), is a sigmoidal curve (see Figure 6-1). Hemoglobin affinity for oxygen is influenced by factors such as pH, temperature, 2,3-diphosphoglycerate (2,3-DPG), and CO2 that can cause a shift in the dissociation curve, making O2 more or less readily released into the tissues.
Defects in Oxygen Delivery
Most forms of shock impair oxygen delivery. Tissue hypoxia caused by impaired oxygen delivery can result from a reduction in cardiac output, a reduction in hemoglobin, a reduction in SaO2, or any combination of these.
Cardiac output is the main determinant of tissue perfusion (i.e., blood flow through peripheral circulation). Hypovolemic, cardiogenic, and distributive shock affect cardiac output and result in abnormalities in tissue perfusion. Tissue hypoxia derived from impairment of tissue perfusion is generally described as ischemic hypoxia. Direct measurements of tissue perfusion are not easily performed in clinical patients. It is understood that impaired cardiac output results in impaired tissue perfusion, but conversely, it is possible to have regional impairments of tissue perfusion without a reduction in cardiac output.
Cardiac output can be impaired as a result of alterations in heart rate or stroke volume. Dysrhythmias that lead to pronounced bradycardia, tachycardia, or disorganization of the cardiac cycle will result in decreased tissue perfusion. Although cardiogenic shock may be the result of a dysrhythmia, all forms of shock may lead to rhythm disturbances (typically ventricular premature beats) that could further impair perfusion.
Stroke volume is influenced by preload, afterload, and contractility. The classic example of a reduction in preload is hemorrhagic shock; loss of whole blood results in absolute hypovolemia and an inability to adequately fill the ventricle (Box 6-2). Hypovolemia can also result from loss of electrolyte-rich fluid (e.g., with profuse vomiting and diarrhea) or loss of pure water (e.g., in heatstroke and adipsia). In addition to external loss, fluids can be sequestered internally in “third spaces,” in which fluid is not available to replenish the intravascular volume. Examples of third-spacing include ascites, pleural effusion, and fluid sequestration in the gastrointestinal tract. End-diastolic volume can also be limited by impaired right ventricular filling. A “tension pneumothorax” increases intrathoracic pressure and results in collapse of the great vessels and right ventricle. Pericardial effusion can lead to cardiac tamponade and collapse of the chambers of the right heart. Restrictive cardiac disease will also impair diastolic function.
Afterload is typically maintained in compensated hypovolemic and cardiogenic shock. The normal response to preserve vital organ perfusion is vasoconstriction of peripheral vascular beds. Clinically, this is apparent as cool extremities, pale mucous membranes, and prolonged capillary refill time. Abnormal (decreased) afterload is the primary mechanism that leads to distributive shock. Circulating mediators, including nitric oxide, prostanoids, and complement, lead to inappropriate vasodilation, reduction of effective circulating volume, hyperemic mucous membranes, and rapid capillary refill time. Impaired contractility can result from primary cardiac disease, drugs, myocardial ischemia (as can occur with persistent tachycardia), or circulating inflammatory mediators. Cytokines such as tumor necrosis factor-alpha (TNF-α) and interleukin (IL)-1-beta exert short-term direct depressant activity over the myocardium and may contribute to cellular signaling events (e.g., apoptosis, nitric oxide pathway activation, calcium homeostasis imbalance, reduced response to catecholamines) that lead to the prolonged cardiac dysfunction seen in sepsis.75
Adequate arterial oxygen content is essential to maintain oxygen delivery. Three main categories of abnormalities can result in reduction of arterial oxygen content: anemia, altered hemoglobin function, and hypoxemia (reduced SpO2 [oxygen saturation of hemoglobin in peripheral blood as measured by pulse oximetry] or reduced SaO2). Blood loss, red cell destruction, or reduced red cell synthesis can affect the amount of hemoglobin in the blood, leading to anemia and decreased arterial oxygen content. Even in the face of adequate amounts of hemoglobin, arterial oxygen content can be reduced as a result of impaired hemoglobin function. Toxins, such as carbon monoxide, can impair the ability of hemoglobin to carry O2, which leads to tissue hypoxia. Shock is typically a state of acidemia that results in a decreased hemoglobin affinity for oxygen, which will decrease the ability of hemoglobin to take up O2 but enhances the release of O2 in the tissues. Hyperthermia, hypercarbia, and increased 2,3-DPG also result in decreased O2 affinity (see Figure 6-1). In profound cases of shock, hypothermia can complicate delivery of oxygen to the tissues because of the increased O2 affinity of hemoglobin. If hemoglobin is adequate and is able to carry and release O2, but arterial oxygen content is low, then impairments in pulmonary function and gas exchange are likely culprits.
Oxygen Uptake
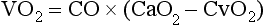
Oxygen uptake (VO2) measures the rate at which oxygen leaves hemoglobin in the systemic capillaries and reaches the tissues. Tissues lack storage systems for oxygen; therefore, global oxygen uptake is equivalent to global oxygen consumption. Oxygen uptake is a function of cardiac output and the difference between arterial oxygen content and venous oxygen content (CvO2) (this relationship is known as the Fick equation). Similar to arterial oxygen content, venous oxygen content is determined by the amount and saturation of hemoglobin and dissolved oxygen in venous blood:
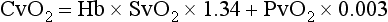
Hemoglobin saturation of venous blood, or SvO2, should be measured on a sample collected from the pulmonary artery (mixed venous oxygen tension [PvO2]). Mixed venous samples are representative of global VO2. Peripheral venous samples reflect the VO2 of the organs being drained and can be highly variable. Although the correlation between mixed venous and central venous (cranial vena cava) hemoglobin saturation is variable, for clinical purposes, mixed venous oxygen saturation is often substituted by central venous oxygen saturation (ScvO2).81
ScvO2 has been recognized as a valuable endpoint of resuscitation.82 If oxygen delivery is adequate, sufficient O2 should remain in the venous blood to provide at least 70% saturation of the hemoglobin. Although low ScvO2 can be attributed to low DO2 and low tissue oxygenation, five commonly described defects occur in VO2 in which the ScvO2 can be adequate, but tissue oxygenation can be impaired (Box 6-3).
Box • 6-3 Defects in Oxygen Uptake
• Diffusional shunting: This is caused by slow blood velocity (prolonged transit time), which favors diffusional oxygen transport over convective oxygen transport (i.e., the movement of oxygen following a concentration gradient prevails over transport of oxygen within the bloodstream). It may occur in the intestine during shock states. The intestine relies on countercurrent blood flow to deliver blood to the villi. If the rate of flow is so slow that oxygen from the arterial blood can diffuse to the venous blood before it is delivered to the tip of the villus, the intestine will be hypoxic, but venous saturation will actually be increased.
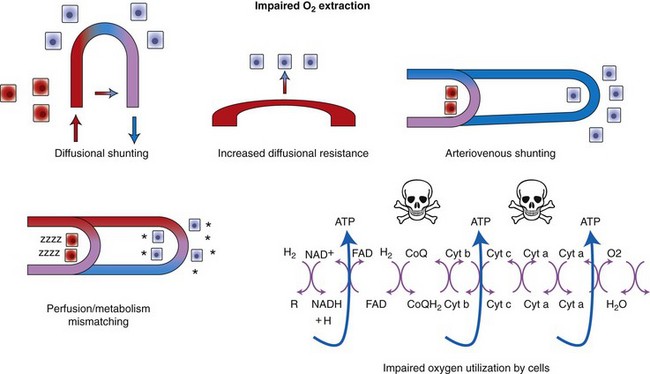
Adapted from Ince C, Sinaasappel M: Microcirculatory oxygenation and shunting in sepsis and shock. Crit Care Med 27:1369, 1999.
• Diffusional resistance: For cells to utilize oxygen, O2 must diffuse from the vascular space through the tissues, into the cell, and finally to the mitochondria. Oxygen is poorly soluble in aqueous solutions; therefore tissue edema will increase diffusional distance and limit oxygen availability to the tissues. Hypoxia may increase edema by increasing vascular permeability, thus leading to a “feed-forward” cycle of hypoxia and edema and more hypoxia. Impaired diffusion will result in a smaller gradient, less driving pressure for the oxygen to leave the hemoglobin, and thus higher venous saturation, despite tissue hypoxia.
• Arteriovenous shunting: One of the most obvious reasons for failure of oxygen uptake by the cells is the loss of normal capillary flow. In sepsis, trauma, and diseases associated with systemic activation of the inflammatory response, capillary obstruction can result from neutrophil-endothelial interaction, platelet aggregation, thrombi, or loss of normal vasodilatory function. As a result, arterial blood bypasses the hypoxic bed, leading to an increase in venous oxygen saturation.
• Perfusion/metabolism mismatch: A cause of inadequate tissue oxygenation is perfusion/metabolism mismatch. Despite the normal excess (approximately four-fold) of oxygen delivery in relation to tissue demand, in some tissues and in some disease states the increased metabolic demand combined with reduced blood flow can lead to tissue hypoxia. A classic example is sustained tachycardia, when coronary perfusion is reduced because of shortened diastole, yet the metabolic demand of the myocytes is increased.
• Cytopathic hypoxia (mitochondrial dysfunction): Cytopathic hypoxia is classically associated with sepsis. Experimental studies of sepsis have demonstrated that despite direct measurement of adequate cellular oxygen, mitochondrial respiration is impaired. Proposed mechanisms to explain this “cytopathic hypoxia” include the following:
Oxygen Extraction Ratio
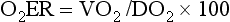
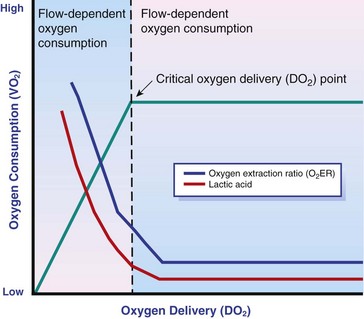
Oxygen extraction ratio (O2ER) is the ratio between oxygen uptake and oxygen delivery expressed as a percentage; it represents an index of the efficacy of tissue extraction of oxygen from the capillary bed. Oxygen extraction ratios of different organs are extremely variable in physiologic and pathologic conditions. Global O2ER under resting physiologic conditions is approximately 20% (20.5 ± 5.7 in dogs)36 but can increase to 60% to 70% in cases of increased metabolic demand and/or a reduction in oxygen delivery.102 Variation in O2ER represents an adaptive system that allows metabolism to maintain a constant VO2 over a wide range of DO2 variability without compromising aerobic metabolism.
DO2/VO2 Curve
Oxygen delivery and uptake are interrelated through a bi-linear relationship (Figure 6-2).47 Over a wide range of values, VO2 remains constant and independent of DO2. This relationship corresponds to the plateau of the graph and is defined as O2 supply–independent VO2. The plateau is not completely flat because the metabolism of some organs, such as the kidney, is partially flow dependent. As oxygen supply decreases, VO2 remains stable initially as a result of increased oxygen extraction. However, below a critical oxygen delivery (cDO2), the increase in oxygen extraction is no longer able to match aerobic metabolism requirements, and oxygen consumption becomes dependent on its supply (supply-dependent VO2). cDO2 is known as the anaerobic threshold, because at cDO2, energy production through oxidative pathways is limited, prompting a shift toward a much less efficient anaerobic metabolism.
Cellular Response to Hypoxia and Lactate
During hypoxia, lack of oxygen leads to an upstream blockage in the mitochondrial electron transport chain, preventing the entry of pyruvate into the Krebs cycle. In this setting, cell energy production relies exclusively on anaerobic glycolysis. To avoid the accumulation of byproducts that would slow the glycolytic reaction, pyruvate is converted to lactate by the enzyme lactate dehydrogenase (LDH). This reaction regenerates NAD+, a necessary coenzyme for continued anaerobic glycolysis, thereby preserving adenosine triphosphate (ATP) production. When oxygen is present, lactate can be converted back to pyruvate or glucose and used as a source of energy in several organs, including heart, brain, liver, and skeletal muscle. However, if the hypoxia is global, lactate will diffuse into the bloodstream. Major sources of lactate are muscle and the gastrointestinal tract. Hyperlactatemia occurs when lactate generation exceeds its clearance by metabolism, primarily in the liver and kidneys.4 Hyperlactatemia induced by tissue hypoxia is commonly associated with metabolic acidosis. However, lactate production from glucose does not generate hydrogen ions (H+). During anaerobic metabolism, concurrent accumulation of H+ is derived from changes in other metabolic pathways. The degradation of ATP to adenosine diphosphate (ADP) produces H+ that under aerobic conditions are consumed by mitochondrial ATP production. In anaerobic conditions, these H+ accumulate in parallel with lactate, creating an acidemic state commonly referred as metabolic lactic acidosis.54
Inflammation in shock can be triggered by the inciting cause of shock, such as bacterial components in septic shock. Other forms of shock also trigger inflammatory responses through hypoxia. Cell death from energy failure and loss of electrical and chemical gradients will result in the release of endogenous danger signals (also referred to as danger-associated molecular patterns, or DAMPs).52 These danger signals are typically intracellular components that when released into the extracellular space bind to receptors on cells of the innate immune system and trigger an inflammatory response. In addition, exposure of cells to hypoxia can initiate activation of transcription factors (i.e., hypoxia-inducible factors; Box 6-4) that are responsible for the adaptive response to hypoxia, as well as activation of inflammatory mediators. Another cause of inflammation in shock is the reintroduction of oxygen to previously hypoxic or ischemic tissues. This reperfusion injury is associated with the generation of damaging oxygen and nitrogen free radicals. When tissues are ischemic, blood flow is compromised, resulting in decreased delivery of oxygen and nutrients. Ischemic tissues become hypoxic, consume the available nutrients, and accumulate waste products like lactate. The energy supply in the form of ATP is depleted, and the byproducts, inosine and hypoxanthine, accumulate. Intracellular calcium increases, which results in the activation of calpain, a proteolytic enzyme. The enzyme that normally metabolizes hypoxanthine, xanthine dehydrogenase, is converted by calpain to xanthine oxidase. During the ischemic phase, hypoxanthine accumulates because xanthine oxidase requires molecular oxygen. With the reintroduction of oxygen, xanthine oxidase metabolizes hypoxanthine and generates superoxide, a potent free radical. In addition, during ischemia, the cell responds to hypoxia and switches from aerobic to anaerobic metabolism through the activation of hypoxia-inducible factors (see Box 6-4). In addition to the compensatory mechanisms that are activated, several inflammatory mediators are induced by hypoxia. Nitric oxide synthase, which also requires oxygen for the production of nitric oxide nitric oxide, accumulates, and when oxygen is reintroduced, nitric oxide production can occur rapidly. Nitric oxide, a reactive nitrogen species, can rapidly interact with superoxide and form peroxynitrite. Upon reperfusion, the production of reactive oxygen and nitrogen species causes tissue damage and the release of increased numbers of cytokines and other inflammatory mediators. Neutrophils and endothelial cells are activated, further amplifying the damage and obstructing capillaries and causing a “no reflow” phenomenon that prevents normal perfusion.58,60
Hemostatic derangements play a major role in the pathophysiology of shock, independent of the inciting cause of shock. Altered coagulation appears early in the course of shock, especially when shock is caused by substantial tissue injury (trauma) or a systemic inflammatory process (sepsis). Several mechanisms contribute to hemostatic derangements37:
• Tissue trauma favors coagulation and concurrent fibrinolysis. Endothelial damage and exposure of subendothelial collagen and tissue factor activate the coagulation cascade. Endothelial damage also induces a hyperfibrinolytic state due to increased synthesis and release of tissue plasminogen activator (tPA).
• Shock, through hypoperfusion, enhances the effects of tissue trauma and promotes an anticoagulant and hyperfibrinolytic state. These hemostatic derangements result from ischemia-induced endothelial release of tPA, inhibition of plasminogen activator inhibitor (PAI)-1, and increased activation of protein C. Tissue trauma AND shock are the main contributors to coagulopathy in the early phases of shock.
• Inflammation may be caused by the inciting cause of shock (sepsis) or may appear later in the course of shock as a consequence of tissue hypoxia and ischemia-reperfusion injury. Inflammation and coagulation are closely interrelated by a bidirectional cross-talk.48 Proinflammatory cytokines induce the expression of tissue factor by mononuclear cells and may directly activate platelets, causing a systemic procoagulant effect. Inflammation contributes to activate coagulation, also through the downregulation of all major anticoagulant pathways (i.e., antithrombin, activated protein C, and tissue factor pathway inhibitor). Coagulation factors (e.g., thrombin) and anticoagulant factors (e.g., activated protein C) may, in turn, affect inflammation by interacting with endothelial and mononuclear cell receptors and modulating cytokine expression and cellular interactions. Systemic inflammation induces a prothrombotic state and eventually will lead to disseminated intravascular coagulation (DIC).
• With the evolution of shock, other factors aggravate the coagulopathy. Hypothermia is common in advanced stages of shock and inhibits platelet aggregation and coagulation factor activity. Acidosis, associated with tissue hypoperfusion, impairs coagulation factor activity. Dilution of coagulation factors may occur as a result of fluid shifts and fluid therapy during resuscitation. Moreover, some resuscitative fluids (synthetic colloids) can directly interfere with hemostasis.
Pathophysiology of Shock
Hemodynamic and metabolic derangements associated with shock are sensed by several different mechanisms that activate a compensatory response. The goal of this response—to support vital organs with adequate oxygen delivery—is achieved through several different mechanisms44:
• Maintaining mean circulatory pressure (circulating volume and pressure)
• Maximizing cardiac performance
The reduction in circulating volume is sensed by baroreceptors located in large thoracic arteries (i.e., carotid sinus and aortic arch) and low-pressure stretch receptors in the atria and pulmonary arteries. Stimulation of these receptors initiates the immediate (within seconds) activation of strong sympathetic reflexes that cause the following:
• Constriction of most of the arterioles, leading to an increase in systemic vascular resistance and redirection of blood flow toward vital organs
• Constriction of large capacitance venules and veins, resulting in an increase in venous return and a reduction in the effective vascular volume (in normal conditions, reservoir veins contain 60% of the circulating volume)
• Marked increase in heart rate, with a consequent increase in cardiac output
Under these conditions, blood flow is selectively redirected toward vital organs, which maintain flow through an autoregulatory system that is relatively independent of sympathetic discharge and blood pressure. These protective mechanisms aim to preserve adequate perfusion, especially of cerebral, coronary, and renal circulation. This selective increase in vascular resistance occurs at the expense of cutaneous and splanchnic circulation and contributes to some of the clinical signs of shock, including pale mucous membranes, increased capillary refill time, and cool extremities.
Chemoreceptors, strictly associated with baroreceptors and located in carotid and aortic bodies, activate the sympathetic nervous system in response to a reduction in O2 and, to a lesser extent, an increase in circulating H+ or CO2. Chemoreceptors in the brainstem are much more sensitive to H+ and CO2. Peripheral chemoreceptors use the same pathways as baroreceptors but play a more important role in the regulation of ventilation. Chemoreceptor activation induces an increase in respiratory rate with enhanced oxygenation and CO2 elimination.
Sympathetic activation is enhanced by a neuroendocrine response with the release of angiotensin and vasopressin (AVP; also called antidiuretic hormone, or ADH). Angiotensin is produced during hypovolemia through activation of the renal renin-angiotensin-aldosterone system in response to decreased stretching of glomerular afferent arteries and reduced sodium delivery at the level of the macula densa. Moreover, the renin-angiotensin-aldosterone system can be directly activated by sympathetic stimulation. Renin converts angiotensinogen to angiotensin I, which is subsequently converted to angiotensin II, a potent vasoconstrictor and stimulus for Na+ and water retention in the kidney. Angiotensin II also promotes the secretion of aldosterone, which further increases sodium retention.
Vasopressin is a hormone that is synthesized in the hypothalamus and released at the level of the posterior pituitary gland. In physiologic conditions, it acts in the collecting duct of the kidney to retain water. Vasopressin secretion is regulated by plasma osmolarity as sensed by hypothalamic osmoreceptors. During hypovolemia, vasopressin release is also triggered by a decrease in blood pressure or blood volume mediated by the activation of low-pressure stretch receptors and baroreceptors. In these conditions, vasopressin not only increases water retention, but exerts a powerful vasoconstrictor effect.
As a part of the neuroendocrine response to shock, the sympathetic nervous system also activates the hypothalamus-pituitary-adrenal axis with the secretion of corticotropin-releasing hormone (CRH) from the hypothalamus, adrenocorticotropic hormone (ACTH) from the pituitary, and eventually cortisol from the adrenal gland. Cortisol, along with growth hormone (also released from the pituitary), epinephrine, and glucagon (from sympathetic stimulation of the pancreas), promotes a shift in metabolism toward a catabolic state, increasing extracellular glucose and facilitating readily available energy for the “fight or flight” response.
The decrease in mean arterial pressure, along with constriction of precapillary arterioles, leads to a fall in hydrostatic pressure in capillaries, with a net shift of body fluids from the interstitium into the intravascular space, augmenting the circulating volume. This compensatory mechanism is a major reason for the observed decrease in total protein and hematocrit (Hct) that can be observed acutely in patients with hemorrhagic shock. In the dog, the decrease in Hct is less predictable compared with the total protein, because the sympathetic drive induces splenic contraction and release of a large volume of red blood cells into the systemic circulation. A typical case of acute hemorrhagic shock may have a packed cell volume (PCV) of 35% to 40% with total protein between 4.5 and 5.0 g/dL on presentation.
Several local mechanisms improve oxygen availability at the capillary level during shock. Tissue hypoxia causes a reduction in cellular PO2, increasing the pressure gradient that drives O2 diffusion. Moreover, the development of local acidosis, along with increased 2,3-DPG inside red blood cells, causes a decreased hemoglobin-oxygen affinity (hemoglobin dissociation curve shifted rightward), enhancing O2 unloading in the tissues.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


