18 Therapeutic Drug Monitoring
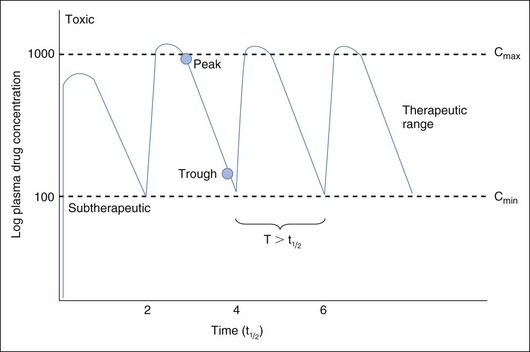
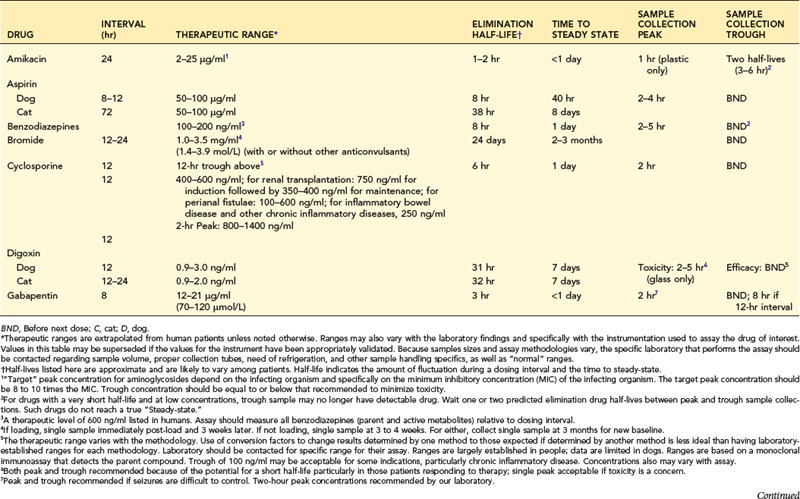
Fixed dosing regimens provided on drug labels are designed to generate plasma drug concentrations (PDCs) within a therapeutic range (Figure 18-1). PDCs are intended to remain above a minimum effective concentration (Cmin) to avoid therapeutic failure while remaining below the maximum concentration (Cmax) and minimizing side effects. Fixed dosing regimens are based on pharmacokinetic studies conducted in a small sample population of normal adults. These regimens, however, are generally administered to unhealthy animals for which drug absorption, distribution, metabolism, or excretion (or a combination of these) have been altered by physiologic factors (e.g., age, gender), pathologic factors (leading to renal or hepatic impairment), or pharmacologic factors (e.g., drug interactions). Furthur, many drugs used in animals are human drugs based on anecdotal doses. As a result, PDC may be higher or lower than expected, increasing risk of toxicity or therapeutic failure, respectively. In these instances, individual monitoring and adjustment of doses using therapeutic drug monitoring (TDM) can optimize drug efficacy and safety. Drugs for which TDM has proven useful in veterinary medicine include several anticonvulsants (e.g., selected benzodiazepines, phenobarbital, primidone, levetiracetam, potassium bromide, zonisamide, gabapentin); antimicrobials (e.g., aminoglycosides: gentamicin, amikacin); cardioactive drugs (e.g., digoxin, procainamide, lidocaine); theophylline; thyroid hormones (for thyroid supplementation); and immunomodulators (e.g., cyclosporine) (Table 18-1).

Implementing Therapeutic Drug Monitoring
Steady State
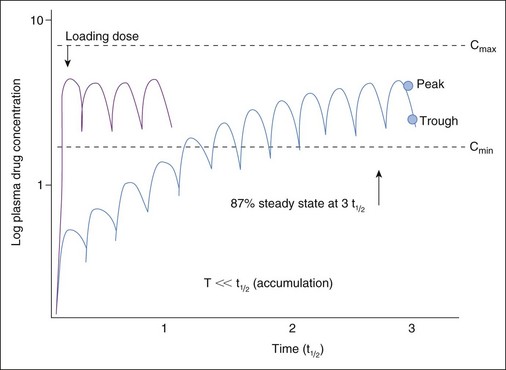
When therapy is initiated, repeated and regular administration of successive doses may result in accumulation of drug. The longer the half-life compared with the dosing interval, the greater the amount of drug accumulation. Eventually the amount of drug eliminated during each dosing interval equals the amount administered with each dose. At this time, steady-state equilibrium has been reached; peak (i.e., the highest blood concentration of the drug) and trough (i.e., the lowest blood concentration of the drug) blood concentrations remain constant across multiple dosing intervals as long as neither the dose nor interval is changed. Ideally, TDM and clinical response to therapy should be implemented at steady state to assess maximum response. Time taken for steady state to be reached depends only on the rate of drug elimination (i.e., drug elimination half-life), irrespective of dose or interval. With multiple dosing of drugs that accumulate, PDC reaches 50% of steady-state concentrations at one half-life (t1/2), 75% by two half-lives, 87.5% at three half-lives, and so forth (Figure 18-2). Generally, TDM should not be implemented until three to five half-lives have elapsed since initiation of drug therapy at the same dosing regimen. For example, for phenobarbital with a t1/2 of 72 hours, TDM should not be implemented until approximately 15 days after initiation of therapy. If any aspect of the dosing regimen is changed, the same time period (i.e., five half-lives) must elapse for steady-state plasma concentrations to be re-established. An exception might be made for bromide, for which a sample might be taken at one half-life (i.e., 3 weeks) after a dose is begun to proactively assess a dosing regimen; at one half-life, drug concentrations should approximate the steady-state concentration. Times taken to achieve steady state (as well as other pharmacokinetic and dosing information) are listed in Table 18-1. However, these times are based on average half-lives determined from sample populations. Some drugs (e.g., zonisamide, cyclosporine) have half-lives that vary dramatically between individuals. Drug interactions may cause similar variations. Time to steady state of a drug in an individual patient can be determined by measuring the half-life of a drug in that patient (i.e., by collecting both a peak and trough sample).
Loading Dose
When the clinical situation necessitates immediate attainment of therapeutic drug concentrations (e.g., a seizuring patient being treated with bromide), PDC predicted at steady state can be achieved more rapidly by administration of a single loading dose followed by recommended maintenance doses (see Figure 18-2). Loading doses are based on the volume of tissue that dilutes the drug; bioavailability of the drug also affects loading dose for those drugs not administered intravenously (IV). Despite the fact that predicted steady-state PDCs have been achieved by loading, PDCs are not yet at steady state and will not be until equilibrium has been reached at three to five half-lives of the drug at maintenance dose in the patient. PDC achieved after loading may increase or decrease if the maintenance dose is more or less, respectively, than that eliminated during each dosing interval. For bromide in particular, TDM should occur within 1 to 3 days after loading to confirm that targeted concentrations (i.e., predicted steady-state concentrations) have been achieved and again one drug half-life later (i.e., 3 to 4 weeks for bromide) to ensure that the maintenance dose is maintaining PDC achieved with loading. Although use of a loading dose decreases the time taken for maximum response to occur (by avoiding slow accumulation to steady state), hazards of adverse reactions are much greater. Thus loading doses are not advised for drugs characterized by a narrow therapeutic index and that tend to cause undesirable adverse reactions (e.g., digoxin).
Number of Samples
The relationship between drug half-life and dosing interval (as well as the intent of monitoring) determines the number of samples to be collected. If the dosing interval is longer than drug half-life (e.g., diazepam, levetiracetam, gabapentin, most antibiotics, cyclosporine), the PDC fluctuates widely during each dosing interval (see Figure 18-1). Assuming that both efficacy and safety are of interest, one should collect two samples to coincide with peak and trough concentrations. For drugs with a long half-life compared with dosing interval, drug concentrations do not change much during each dosing interval, and a single sample generally reflects PDC throughout the dosing interval. The need for one or two samples varies for some drugs. For digoxin, collection of both a peak and trough sample might be considered because of the narrow therapeutic index of this drug and marked variability in half-life that can occur in patients receiving cardiovascular drugs. A single trough sample is often sufficient for phenobarbital; however, because elimination half-lives of less than 24 hours have been measured, clinicians should collect both a peak and trough sample in patients in which seizure control is difficult. Although cyclosporine has a short half-life, recommendations extrapolated from humans may be based on either a peak or trough sample, or both.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


