Chapter 33 The Genus Clostridium
Clostridia are large, sporulating, gram-positive, oxygen-tolerant to strictly anaerobic rods. Clostridium piliforme is the exception, in that it is gram negative. The source of clostridia can be exogenous (most commonly the soil) or endogenous (often from the intestinal tract). Most pathogenic clostridia produce one or more toxins, and direct or indirect evidence links many of these with pathogenesis (Table 33-1). Myonecrosis often follows mechanical injury, with clostridial spores germinating in damaged, ischemic muscle. Toxemia can also ensue when spores are deposited in wounds, or when spore-containing tissues are damaged. Disruption of normal flora by antibiotic therapy or sudden dietary changes may precede enteric infection, which is frequently accompanied by intestinal lesions; in some types of enterotoxemias, intestinal lesions are minimal. Disease can also result from ingestion of preformed clostridial toxins. Death is a frequent endpoint, and is often acute or peracute. Vaccination with bacterin-toxoids or toxoids is an effective prophylactic measure for many clostridial diseases.
TABLE 33-1 Animal Pathogenic Clostridia
| Pathotype | Species | Disease |
|---|---|---|
| Neurotoxic | C. botulinum | Botulism |
| C. tetani | Tetanus | |
| Histotoxic | C. perfringens | Myonecrosis, gas gangrene |
| C. septicum | Malignant edema, braxy | |
| C. chauvoei | Blackleg | |
| C. novyi | Myonecrosis, infectious necrotic hepatitis (black disease), bacillary hemoglobinuria (redwater) | |
| C. sordellii | Myonecrosis, enteritis | |
| Enteric | C. perfringens | Enteritis, enterotoxemia |
| C. difficile | Diarrhea, antibiotic-associated diarrhea, pseudomembranous colitis, colitis X | |
| C. colinum | Quail disease | |
| C. spiroforme | ı-Enterotoxemia | |
| C. piliforme | Tyzzer’s disease |
Animal pathogenic clostridia can be conveniently categorized as neurotoxic, enteric, and histotoxic (see Table 33-1). Some (e.g., Clostridium perfringens) are heterogeneous and thus rather cosmopolitan in terms of hosts and systems affected, whereas others (e.g., Clostridium colinum) cause a single, well-defined syndrome.
NEUROTOXIC CLOSTRIDIA
Diseases and Epidemiology
After germination in the wound, C. tetani produces a neurotoxin that causes the majority of the symptoms of tetanus. The incubation period ranges from 24 hours to 2 weeks, varying with the toxogenicity of the infecting strain, the rate at which toxin is transferred to target tissues, and host sensitivity. Ascending tetanus follows retrograde, intraaxonal transport of toxin along the peripheral motor nerves to the central nervous system (CNS). Toxin crosses the synapse and binds to presynaptic axonal terminals, causing motoneuron hyperactivity, with sustained spasms inthe innervated muscles. Other muscle groups are affected when toxin travels within the spinal cord. Descending tetanus results from vascular dissemination of toxin, and clinical effects often begin in sites distant from the infection.
Outbreaks of type C botulism in waterfowl may originate with, and be sustained by, toxin in tissues of dead invertebrates. Invertebrate larvae ingest toxin from vertebrate carcasses and are consumed by fowl. Sporadic outbreaks of type E botulism in wild birds may be associated with consumption of toxin-bearing fish. Dabbling ducks are commonly affected, as are shorebirds. Mortality in migratory populations can exceed 50,000 birds in a single season.
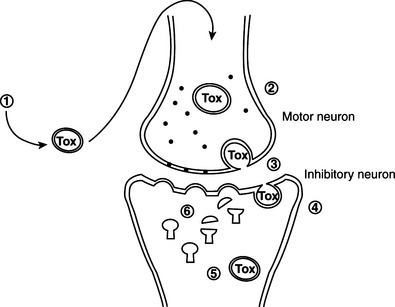
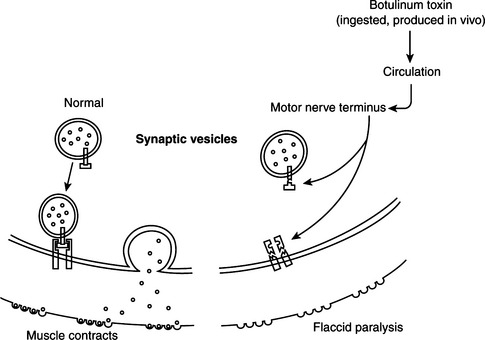
Pathogenesis of tetanus and botulism.
Tetanus neurotoxin and botulinum neurotoxins serotypes B, D, F, and G cleave VAMP/synaptobrevin, a membrane protein of the synaptic vesicles (Figures 33-1 and 33-2). Cleavage is atdifferent, single sites for each toxin. Botulinum A and E neurotoxins cleave synaptosomal-associated protein–25 (SNAP-25), a component of the presynaptic membrane, at two different carboxyl-terminal peptide bonds. Serotype C specifically cleaves syntaxin, another protein of the nerve plasmalemma. These three proteins are conserved from yeast to humans and are essential in a variety of docking and fusion events in every cell. The end result of toxin action on target cells is blockage of acetylcholine release, and muscle paralysis.
HISTOTOXIC CLOSTRIDIA
Diseases, Epidemiology, and Pathogenesis
Histotoxic clostridia are common pathogens of humans and domestic animals, and a limited group causes most of the infections (Table 33-2). The hallmark is enthusiastic toxinogenesis, but common themes are acquisition of the infecting organism from soil or an endogenous source (such as the intestinal tract), entry to tissue following trauma, local multiplication and toxin production, occurrence of extensive local (and often systemic) tissue damage, and rapid death of the host. In animals, control by vaccination has decreased the incidence, and perhaps also the visibility, of some clostridial diseases, but renewed interest in mechanisms of pathogenesis, and to some degree the continuing interest inprevention of battlefield clostridial infections or intoxications, has yielded new information, particularly about modes of toxin action.

Clostridium septicum.
Malignant edema in domestic animals(Figure 33-3) usually follows direct contamination of a traumatic wound. Genital infectionscan be associated with mismanaged attempts at delivery, and umbilical infections are not infrequent in lambs. Hemorrhage, edema, and necrosis spread rapidly along fascial planes from the point of infection. As in human infections, the developing lesion is initially painful, warm, and it pits on pressure, but gradually becomes crepitant and cold, with loss of feeling. Death follows a period of fever, anorexia, and depression, often in less than 24 hours.

FIGURE 33-3 Clostridium septicum–associated necrotic myositis (malignant edema).
(Courtesy Raymond E. Reed.)
Clostridium novyi.
Bacillary hemoglobinuria is most common in well-nourished animals more than 1 year of age. Deposition of type D spores or vegetative cells in the digestive tract and liver follows ingestion. Immature flukes migrate through the liver, causing hepatic necrosis and hypoxia, and inducing germination of spores in Kupffer cells. β-Toxin causes hepatic necrosis, and dissemination through the bloodstream leads to intravascular hemolysis and hemorrhage. Fever, pale mucous membranes, anorexia, abdominal pain, and hemoglobinuria (from which the common name “redwater” is derived) are typical clinical signs; when hemoglobinuria appears, 40% to 50% of red cells have been lysed, and death ultimately results from anoxia. Serosal effusions and a large circumscribed liver infarct are pathognomonic, and gram-positive rods are abundant in the sinusoids. Thus the pathogenesis of bacillary hemoglobinuria is similar to that of black disease of sheep, except that the primary toxin is β- rather than α-toxin. A typical case fatality rate is 90%to 95%.
Type B may be involved in an emerging problem with sudden death in sows (Figure 33-4), which is associated with multiplication of the organism in the liver at or about the time of parturition. This disease has not been reproduced by experimental inoculation of sows.
FIGURE 33-4 “Aerochocolate” liver from a sow with Clostridium novyi type B infection.
(Courtesy Raymond E. Reed.)
Clostridium chauvoei.
Blackleg (Figure 33-5) occurs most commonly in well-fed cattle less than 3 years of age. Lesions usually occur in hindlimb muscle mass, but may be seen only in myocardium, or possibly the diaphragm or tongue. In sheep the diseasemore frequently presents as a wound infection resembling malignant edema (C. septicum infection) or gas gangrene (C. perfringens infection). Clinical signs include high fever, anorexia, depression, and lameness. Many lesions are internal, but superficial ones may be crepitant on palpation. Sudden death, without observed clinical signs, is common.
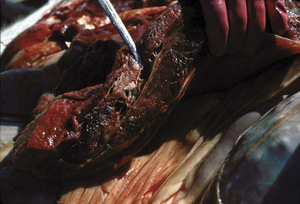
Ingestion is the most probable route ofexposure in cattle, and various tissues, especially skeletal muscle, are seeded with spores. Outbreaks may be due to spread of infection or a common source of local muscle anoxia, such as overexercise induced by encounters with phlebotomous insects. Damage to the muscle provides conditions favoring germination of dormant spores, with subsequent multiplication and toxin production. Acute indigestion may also initiate these events.
Clostridium piliforme.
The etiologic agent of Tyzzer’s disease is Clostridium piliforme (Figure 33-6). Formerly known as Bacillus piliformis, recent studies of the 16S rRNA sequence have yielded a phylogenetic tree that suggests thatthe organism’s closest relatives are Clostridium coccoides, Clostridium oroticum, Clostridium clostridiiforme, Clostridium symbiosum, and Clostridium aminovalericum. It has never been cultivated in cell-free medium, but has been propagated in a mouse-embryo fibroblast cell line.
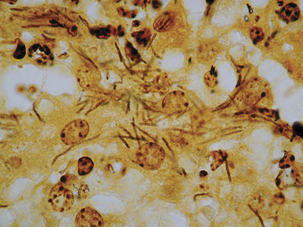
Acutely affected, recently weaned rabbitsmanifest profuse watery diarrhea, with mortality ranging from 15% to 50%. Survivors may be chronically affected, with depression, anorexia, weight loss, and cachexia. Necrotic and hemorrhagic enteritis are observed mainly in the ileum, cecum, and colon, with marked serosal andsubmucosal edema, congestion, and distinctreddening of cecal tissues. Multifocal hepatic necrosis generally appears after the acute phase of the disease in rabbits. Focal myocardial necrosismay also occur. Bundles of slightly gram-negative and silver-positive rod-shaped bacilli can be seen in viable hepatocytes bordering necrotic foci, in myocytes around necrotic foci in the heart, and in enterocytes and smooth muscle cells ofthe muscularis mucosa of the intestine. The occurrence of antibodies to C. piliforme in some species suggests that subclinical infection is common.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


