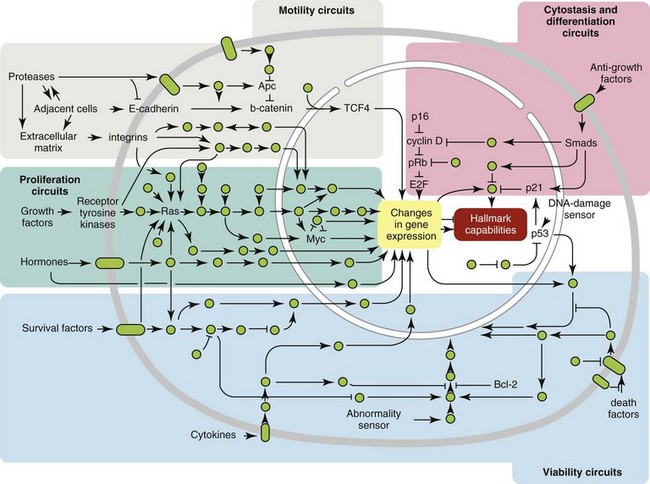
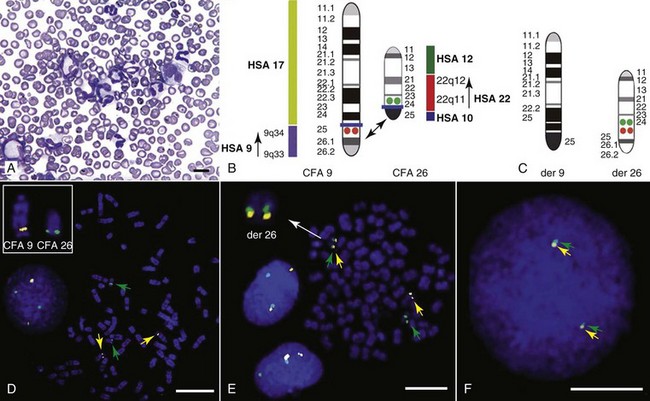
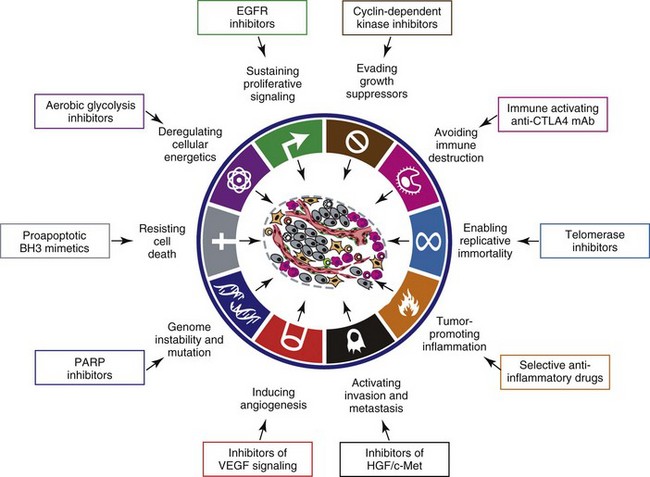
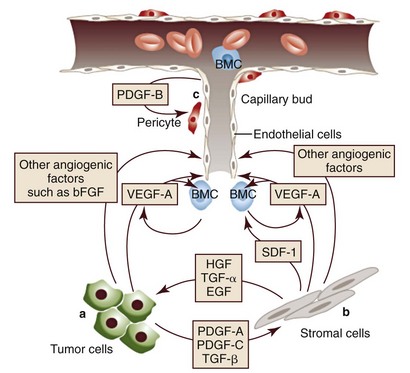
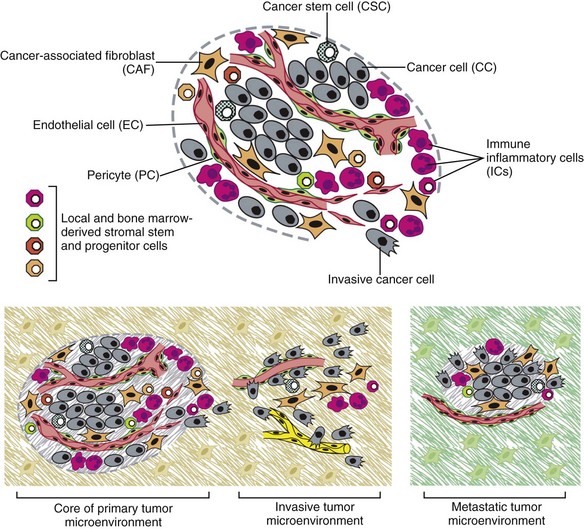
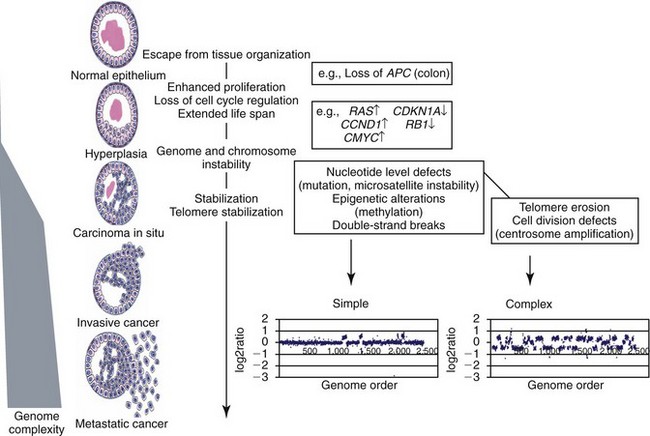
1 This is an exciting and rapidly evolving time in the field of cancer genetics. Although it has been clear for several decades that cancer is a disease driven by the accumulation of genetic abnormalities,1,2 new technologies are rapidly unraveling nuances in how heritable traits influence epigenetics and the tumor environment. Meticulous reductionist research done since the early 1960s helped identify hundreds of genetic abnormalities that are peculiarly associated with specific cancers, and more recent advances allowed for full sequencing of tumor genomes.3–6 The information from these experiments has reinforced current concepts and provided insight into new areas of research. This chapter will focus on contemporary information to provide context for the genetic basis of cancer and how interactions between genes and environment impact the origin, progression, and response to therapy of hematopoietic tumors. It is probably reasonable to say that the next decade will represent a new “golden age” of discovery in cancer genetics, when many of the apparent conflicts from our traditional (reductionist) experimental approaches will be resolved by integration of data from epidemiologic, molecular, and clinical studies into a more holistic understanding of the biology of cancer. The existence of genetic predisposition to cancer is illustrated by well-defined heritable cancer syndromes.7 Over 200 such syndromes have now been defined.8 Even though they account for only 5% to 10% of all human cancers, studies of families with these syndromes provided many of the initial clues to understanding the genetic basis of sporadic (nonheritable) cancers. Although inheritance is recessive, these familial cancer syndromes show dominant patterns of inheritance and many have high penetrance.8 All but two of the known familial cancer syndromes are due to mutations that inactivate tumor suppressor genes. As originally proposed by Knudson in his “two-hit” hypothesis from studies in children with retinoblastoma,9 individuals at risk are obligate heterozygotes (they inherit a mutant allele and a wild-type allele). As it happens, homozygous mutations in critical growth regulatory genes usually cause embryonic lethality; however, in the case in which a single allele is affected, the mutation is present in every cell in the body. Given the rate of spontaneous mutation as described, the probability that the second, wild-type allele will be inactivated in at least one cell is extremely high, therefore facilitating tumorigenesis. This process is called loss of heterozygosity (LOH). A curious observation worth noting is that different mutations in a single gene can predispose individuals to distinct cancer syndromes, whereas independent, single mutations of different genes can result in virtually the same disease, or at least diseases with indistinguishable phenotypes.7 This is not surprising when we consider that commonly affected genes are multifunctional and parts of complex, interactive networks or circuits10,11 (Figure 1-1). Thus a mutation may only alter gene function along one biochemical pathway, leaving its interactions with other pathways intact. Moreover, mutations that contribute to most sporadic cancers are restricted to a small subset of genes,12 many of which also are associated with heritable cancer syndromes. These observations have given rise to competing contemporary theories on the origins of cancer, which are addressed later in this chapter. At least one heritable cancer syndrome (renal carcinoma and nodular dermatofibrosis [RCND] of German shepherd dogs) has been described in dogs.13 The heritable factor (or RCND gene) for this syndrome maps to dog chromosome 5 (CFA 5) and specifically to the folliculin gene, which was recently described as the heritable factor for the corresponding human disease (Birt-Hogg-Dube syndrome).14 It is probable that other syndromes comparable to those that are described in humans will eventually be identified in companion and laboratory animals, but it is unlikely that these will account for more than 5% to 10% of all cancer cases. Unlike diseases due to single gene defects, cancer is a complex, multigenic disease. The “initiation, promotion, and progression” model was among the first to propose a sequential progression of mutations that could account for cancer.15,16 In this model, a genetic event would endow a somatic cell with limitless replicative potential or another growth or survival advantage from other cells in its environment (initiation). Alone, this would not be sufficient to give rise to a tumor, as the cell would remain constrained by environmental factors. A second event would further add to the cell’s ability to outcompete its neighbors in this environment, leading to its potential expansion into a recognizable tumor mass (promotion). Finally, a third event would reinforce the cell’s malignant potential (invasion, tissue destruction, and metastasis), leading to clinical disease (progression). It is important to note that an “event” is not equivalent to a single mutation but rather is more likely to represent a series of mutations that act in concert to alter the cell’s functional and morphologic phenotype. Although this model is overly simplistic and technically flawed (experimental evidence clearly shows that mutations are stochastic and do not occur in step-wise fashion),17 it is nevertheless useful to convey the events that lead to carcinogenesis and it remains the foundation for our current understanding of cancer genetics and cancer evolution. Both the environment and the individual’s peculiar genetic background influence cancer risk and the natural history of tumors. This is especially clear in mice, in which the relative rate of spontaneous cancers and the susceptibility to chemically induced cancers differ according to the genetic background of various inbred strains.18 Similar evidence exists for humans; for example, the risk of habitual smokers to develop lung cancer was found to be tightly linked to a unique allele encoding the alpha-3 subunit of the high affinity nicotinic receptor19–21 and was later extended to this and other loci where additional nicotinic receptor subunits are encoded.22 An association between lung cancer arising from habitual tobacco use and activity of cytochrome P450 enzymes also had been observed repeatedly for many years, and there are indeed P450 alleles (e.g., CYP1B123) that also are associated with lung cancer. Together, these findings illustrate the complex relationship between genetics, environmental exposures, and probability to define cancer risk, prevention, and treatment. Specifically, alleles encoding “higher risk” nicotine receptors appear to modulate nicotine signaling, which in turn is responsible for embedding smoking behaviors and consequently exposure to dozens of potent carcinogens.22,24 Nicotine metabolism itself by P450 enzymes also influences tobacco use; however, conversion of tobacco-specific nitrosamines to mutagenic forms by highly active P450 enzymes modulates risk of transformation.25 Each component of risk is incremental and their interactions are not predictable. It is likely that similar relationships will be operative for many, if not most, common cancers of humans and domestic animals; defining these interactions will be a major emphasis of research during the coming decades. There also is evidence to suggest that in some cases mutations are “directed” due to the presence of a “mutator phenotype,” in which the factors that control DNA replication and repair are prone to more errors than would be expected by simple random events. This leads to different rates of cancer predisposition, which would be higher than the mean in individuals bearing this “mutator phenotype,” and might explain why not all people or animals exposed to similar environmental carcinogens develop the same forms of cancer at the same rate.26 Recent information obtained from massive parallel full genome sequencing of tumor/normal pairs in diffuse large B-cell lymphoma suggests that the mutator phenotypes may be acquired and may involve genes that mediate both DNA repair and chromatin organization.5,6 It is important to note, however, that this does not exclude the possibility that mutator phenotypes also might be heritable. In dogs and other domestic animals, the coexistence of genetic isolates in closed populations we call “breeds,” along with animals of mixed breeding, lends itself to study how a relatively homogeneous background influences cancer in out-bred populations. Preliminary data from whole genome association studies suggest there are distinct heritable traits that segregate with common cancer phenotypes in dogs.27,28 One common finding—and a pervasive obstacle for the completion of these studies—has been the observation of “fixed” risk alleles, making an association between individuals in the breed and disease challenging. Nevertheless, even though at the time of this writing none of these studies were yet published in the peer-reviewed literature, the reader should be alert for upcoming studies that will likely document specific risk genes for histiocytic sarcoma, transitional cell carcinoma, osteosarcoma, hemangiosarcoma, lymphoma, mammary cancer, and melanoma, among others, in susceptible dog breeds, and with the recent advent of the feline genome sequence,29 possibly in specific cat breeds as well.30 It remains to be seen if these traits will be shared between closely related breeds or whether they contribute to risk independently among different breeds.31 Perhaps as important, dogs are the first species in which genetic background has been shown to mold tumor genomes and tumor gene expression profiles.32–35 This knowledge, together with the demonstration that causal, pathognomonic genetic abnormalities are conserved in homologous human and canine cancers,36 opens a new area in which the precise contribution of heritable traits to sporadic cancers can be identified by using comparative systems approaches. These observations also indicate that we must assess “risk” far beyond the conventional idea of “tumor development,” as heritable traits might influence risk by modulating the probability of initiating events, the probability of promoting events, or the probability of progression through the interactions between the tumor and its microenvironment.34 A useful illustration of this concept is the occurrence of prostate cancers in men: virtually all men over 60 years of age will die with prostate cancer, but only a minority will die from prostate cancer, indicating that the major factors that influence the disease are not those that mediate transformation (at least as defined by morphologic appearance and anatomic organization), but instead those that dictate the biologic behavior of the transformed cells in the host. Another important conceptual advance in this regard was the identification that spontaneous canine tumors had highly conserved (homologous) aberrations that had been previously characterized in human tumors. The prototypical example is a structural aberration resulting from a balanced chromosomal translocation that creates a fusion gene comprised of most of the BCR gene (located on chromosome 22 in humans and on chromosome 26 in dogs) and a truncated form of the ABL gene (coincidentally located on chromosome 9 in both humans and dogs) in chronic myelogenous leukemia (CML).36 Both translocations give rise to a derivative chromosome, the Philadelphia (Ph) chromosome in humans and the Raleigh chromosome in dogs, as illustrated by the canine form in Figure 1-2. Numeric aberrations (changes in DNA copy number) are similarly conserved among species, as illustrated by deletions of the RB1 locus, including the associated tumor-suppressing microRNAs in chronic lymphoid leukemias36 and the INK4 locus in T-cell malignancies,37 as well as copy number gains such as Runx2 amplification in osteosarcoma.38 The development of these specific tumors from cells harboring such mutations may not be at all surprising, but why would homologous, highly conserved pathologic rearrangements, deletions, or amplifications occur in cells from organisms separated by more than 40 million years of evolution? Is it possible they are evolutionarily related on a mechanistic basis? For example, rearrangements of the immunoglobulin heavy chain locus and the MYC locus are thought to be due to recognition of MYC flanking sequences by the recombinase enzyme system.36 No such mechanism is known to be operative for other defined sites, so these other mutational events could occur stochastically, with their recurrent characterization across multiple species being the result of the selective advantage provided by the acquired gene to a cell of a highly specific lineage under highly specific conditions. This notion fits with Duesberg’s hypothesis that aneuploidy precedes genetic instability.39,40 Although it is impossible to rule out this argument, the implication would be that such mutational events are phenomenally common, and since they are not otherwise observed frequently in other cells where their occurrence was neutral makes this highly unlikely. Another possibility is that they are related to the nuclear anatomy of the cell and specifically caused by proximity of chromosomal regions, cellular stress, inappropriate DNA repair (or as mentioned previously, recombination), and DNA sequence and chromatin features.41 A third most intriguing possibility is that cellular genomes are reverting to a conformation that was found in a common ancestor (thus the high affinity and specificity between the rearranged chromosomal segments leads to the same recurrent event in many patients) but lost during the process of chromosomal reorganization in evolution, or that these sites represent targets for gene deletions or duplications that have been repeatedly advantageous to species under conditions of natural selection and so have become embedded in their contemporary descendants. The clinical relevance of shared evolutionary and genetic origins should not be lost on physicians, veterinarians, or scientists. Shared origins mean shared biologic behaviors, thus supporting the rationale to apply the same therapies that have been developed for human patients to treat companion animals and vice versa. The best evidence for shared biologic behaviors comes from four recent studies of osteosarcoma, in which gene expression profiling documented overlapping characteristics of this disease in humans and in dogs.35,42–44 Specifically, the use of biased breed cohorts and isolated tumor explants allowed our group to filter genetic noise and stromal signatures to achieve pathologic stratification of osteosarcomas from three independent dog cohorts and five independent human cohorts into prognostically significant groups.35 Thirty years of research culminated in the year 2000 in an insightful and thorough review paper by Douglas Hanahan and Robert Weinberg that synthesized our knowledge into six essential, acquired characteristics necessary for cellular transformation.45 These characteristics included (1) self-sufficiency in growth signals, (2) insensitivity to antigrowth signals, (3) the ability to evade apoptosis, (4) limitless replicative potential, (5) sustained angiogenesis, and (6) the capacity to invade tissues and metastasize. The importance of this paper was less in describing a list of events and more in the synthesis of these events because the concepts proposed by Hanahan and Weinberg created a paradigm shift in our understanding of cancer. Some of the important concepts that were clarified include: No single gene is universally responsible for transformation; five or six mutations are the minimum probable number required to endow the cancer phenotype (an observation that has since been confirmed experimentally3); each step is regulated by multiple interactive biochemical pathways,10 and thus mutations of different genes along a pathway can result in equivalent phenotypes and, conversely, mutations of the same gene can result in different cancers with distinct biology; tumors behave as tissues; and the interactions between the tumor and its microenvironment are major drivers of cancer behavior. In early 2011, Hanahan and Weinberg updated the hallmarks of cancer in a new review that will likely be even more influential than the first.11 In this “next generation,” the hallmarks of cancer were refined and reassessed, and new “enabling characteristics” (genome instability and mutation and tumor-promoting inflammation) and “emerging” hallmarks (deregulating cellular energetics and avoiding immune destruction) were added to the paradigm. The impact of this unifying conceptualization of cancer genetics and this level of understanding are clearly evident when we consider how they have influenced the design, development, implementation, and success of new cancer therapies (Figure 1-3). A summary of the information with added refinements is provided later, and the reader is referred to the original manuscripts for details, since space constraints preclude an extensive review herein. Arguably, the most important event in neoplastic transformation is the capability of cells to sustain chronic proliferation. Under normal conditions, cells communicate with each other and integrate environmental signals by sensing cues and gradients. For example, migration, metabolism, and proliferation of mature hematopoietic cells are regulated in autocrine and paracrine fashions by locally secreted cytokines. The same cytokines may travel systemically and act in an endocrine fashion. Generally, the cytokines work by binding transmembrane receptors, which in turn initiate signaling cascades that culminate in transcriptional changes that allow the cell to adapt its behavior to match the environmental signal. The activity of these cytokines, their receptors, and the corresponding signaling molecules are finely tuned. The system can be shut down when the concentration of the cytokine falls below a threshold that can stably bind the receptor, when the receptor ceases to be expressed, or when signaling molecules are downregulated or otherwise inactivated. However, mutations in even one of the molecules involved in regulating these pathways can provide sustained growth signals in the absence of the initiating cytokine. Among many examples, there is a translocation between chromosome 2 and chromosome 5 (t(2;5)) that is present in almost half of human anaplastic lymphomas. The translocation creates a fusion protein between the nucleophosmin gene (NPM1) and the anaplastic lymphoma kinase gene (ALK), which aberrantly activates the Jak2/STAT5 signaling pathway46 that is normally responsive to various interleukins (IL), including IL-2, IL-3, and IL-6. The genes that encode the normal growth-promoting proteins (such as ALK, Jak2, and STAT5) are called proto-oncogenes; the mutated versions that allow cells to gain self-sufficiency from the environmental signals are called oncogenes. It is important to note that not all growth-promoting genes have the capacity to become oncogenes and that the outcomes of oncogenic activation are most commonly senescence or apoptosis, unless there are additional events that promote stable transformation and survival. In addition to the hallmark capability of inducing and sustaining positively acting growth-stimulatory signals, cancer cells must also circumvent powerful programs that negatively regulate cell proliferation; many of these programs depend on the actions of tumor suppressor genes. To maintain homeostasis, cells also must integrate antigrowth signals from the environment. Quiescence in nonhematopoietic cells is enforced by signals delivered by contact inhibition.47 Hematopoietic cells, on the other hand, utilize cell-cell contacts to maintain interactions within the niche and to regulate the timing and intensity of hematopoiesis, inflammation, and immunity.48 “Stop” signals are usually delivered and integrated by the products of tumor suppressor genes, which derive their name largely from the observation that their inactivation facilitates tumor formation. Tumor suppressor genes balance the activity of growth-promoting proto-oncogenes and tend to act in tandem with these in most biochemical pathways. Loss of function of one or more tumor suppressor genes occurs in virtually every cancer, with inactivation of p53, RB1, PTEN, or CDKN2A each seen in more than 50% of all tumors. Each of these pathways may contribute to the pathogenesis of bone marrow–derived tumors in companion animals, and their dysfunction also may be predictive for outcomes.49–52 Apoptosis, or programmed cell death, is the imprinted outcome for every cell in multicellular organisms. Survival requires support from extrinsic (environmental) factors, as well as precise balance of cellular energetics and metabolism. Bone marrow–derived cells normally undergo apoptosis when concentrations of survival factors (e.g., stem cell factor, IL-3, IL-7) or nutrients are limiting or when there are severe disruptions to cellular bioenergetics.53 A more recent concept in the cell death field is autophagy—a process that tumor cells have efficiently coopted as a means to survive under adverse conditions.54 As part of the autophagy program, intracellular vesicles termed autophagosomes surround intracellular organelles and fuse with lysosomes. There, the organelles are broken down and then are channeled to form new molecules that support the energy-producing machinery of the cell, allowing it to survive in the stressed, nutrient-limited environment that defines most cancers. Tumor cells also must avoid death by anoikis or loss of integral cell-to-cell or cell-to-matrix contacts.47 Absent these physiologic death pathways, the body often reacts to the anatomic and physiologic disruptions caused by cancer cells by targeting these cells for destruction through inflammatory pathways. This is but one pathway that leads to necrosis, since it appears that the process of necrosis also might be regulated genetically, providing another mechanism that favors survival of the whole (organism or tumor) over survival of the one. We are probably on the edge of an explosion of new findings that will further nuance our perception of how evasion (or inciting) of these cell death mechanisms contributes to neoplastic transformation and tumor progression. Immortalization is another essential feature of cancer. The genetic program limits the number of times a cell is able to replicate, the so-called Hayflick limit, and when this limit is reached, replicative senescence is induced. Induction of replicative senescence does not induce death; cells maintain energetic homeostasis and remain functional, but they undergo significant genetic changes characterized by telomere erosion. Cells that are able to replicate must maintain the integrity of telomeres, which are “caps” made of repetitive DNA sequence that protect chromosomes from destruction. Solid tumors acquire immortalization predominantly by activation of the telomerase enzyme system and the consequent maintenance of telomere integrity. In hematopoietic cells, telomerase activity seems to be retained longer than in other somatic cells, so it is possible this facilitates immortalization in lymphoma and leukemia.55 The role of immortalization and the importance of telomerase (both to maintain telomere length and to maintain other biochemical functions that are essential for cell survival) are well established; however, the role of replicative senescence has recently been questioned because improved technology has allowed researchers to circumvent this process in normal cells.11 Mouse models complicate the story due to significant differences in telomere length between rodents and humans, so this is an area in which other models such as companion animals might provide clarity in the future.56 The process of angiogenesis requires the coordinated action of a variety of growth factors and cell-adhesion molecules in endothelial and stromal cells. So far, vascular endothelial growth factor-A (VEGF) and its receptors comprise the best-characterized signaling pathway in tumor angiogenesis.57 VEGF binds several receptor tyrosine kinases, including VEGF receptor-1 (VEGFR-1 [Flt-1]) and VEGFR-2 (KDR, Flk-1). VEGFR-2 is the major mediator of the angiogenic effects of VEGF. However, VEGFR-1 is expressed by some tumor cells and may mediate chemotactic signals, thus potentially having a role in cancer growth. The expression of VEGF is upregulated by hypoxia and inflammation. The transcription factor hypoxia-inducible factor-1α (HIF), which is part of a pathway that also includes regulation by the von Hippel-Lindau (VHL) tumor suppressor gene, is a major regulator of VEGF expression. Under conditions of normal oxygen tension, VHL protein targets HIF for degradation; under low oxygen conditions, HIF increases as VHL-mediated degradation is reduced, allowing for upregulation of VEGF. Other signaling molecules also contribute to angiogenesis, including platelet-derived growth factor-β (PDGF-β) and its receptor (PDGFR), and the angiopoietins. PDGF-β is required for recruitment of pericytes and maturation of new capillaries. Recent studies also document the importance of tumor-derived PDGF in recruitment of stroma that produces VEGF and other angiogenic factors (Figure 1-4). Folkman proposed a role for angiogenesis in cancer more than 30 years ago,58,59 but this idea took time to gain traction in the scientific community. Even after the importance of angiogenesis was recognized, the prediction was that this process would impact solid tumors but would be relatively unimportant for tumors of the blood and lymph (lymphoma, leukemia, and multiple myeloma). The first clues that this notion was mistaken came from unexpected benefits of patients with chronic lymphocytic leukemia (CLL) treated with antiangiogenic compounds,60–62 followed by similar success for some patients with multiple myeloma.63,64 More recently, a European study showed that different histologic types of human non-Hodgkin’s lymphoma show different patterns of angiogenesis, and these can predict outcomes for some of the most aggressive tumors, such as peripheral T-cell lymphomas.65 One study has shown that microvessel density is similarly correlated with the aggressive behavior of canine lymphoma,66 and similar findings have been reported for other blood-derived and solid tumors of dogs, such as mast cell cancer and mammary cancer.67,68 The concepts of how neoangiogenesis contributes to cancer also are undergoing refinement. For example, it is apparent now that clinical trials of antiangiogenic drugs have largely failed because their design was based on incomplete knowledge and thus incorrect assumptions. Perhaps the most informative example is the history of Bevacizumab (a humanized anti-VEGF antibody), which received approval to treat various cancers between 2004 and 2010 after it was shown to confer improved quality of life, albeit with modest survival benefits for patients. On December 6, 2010, the Federal Drug Administration (FDA) issued a press release (http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm237172.htm) that announced the withdrawal of the indication for bevacizumab in treatment of metastatic breast cancer, stating that “… the drug has not been shown to be safe and effective for that use.” The FDA press release also stated, “The agency is making this recommendation after reviewing the results of four clinical studies of Avastin (bevacizumab) in women with breast cancer and determining that the data indicate that the drug does not prolong overall survival in breast cancer patients or provide a sufficient benefit in slowing disease progression to outweigh the significant risk to patients. These risks include severe high blood pressure; bleeding and hemorrhage; the development of perforations (or ‘holes’) in the body, including in the nose, stomach, and intestines; and heart attack or heart failure.” The role of genetic events in invasion and metastasis is still incompletely understood. The classic model of metastasis proposed by Fidler suggests a step-wise acquisition of assets that enables cells to leave the primary tumor site, travel through blood or lymph, invade stroma in favorable locations, and thus become reestablished at distant sites.69 More recent work suggests that most tumors possess the ability to dislodge cells that travel to distant sites, and the ability of such cells to survive in capillary beds may be the most important step in the metastatic process.70–74 In epithelial neoplasms that account for the majority of tumors in humans, the epithelial-to-mesenchymal transition (EMT) has received increasing attention for its role in metastasis. EMT is a developmental program that progenitor stem cells use during morphogenesis and can be physiologically reactivated during wound healing. It remains unclear whether EMT is equally (or less) important in sarcomas more commonly seen in domestic animals, in which the cells of origin seem to retain EMT capabilities to a greater extent. Similarly, there is increasing evidence of interactions between cancer cells, the “initiating” population in the tumor (cancer stem cells [CSCs]), mesenchymal stem cells (MSCs), tumor-associated fibroblasts, inflammatory cells, and angiogenic cells, which may be responsible for invasive behaviors and possibly for survival in hostile environments that exist at distant sites (Figure 1-5). There is a bidirectional flow of information between the tumor and the microenvironment, with each helping to mold the other into functional growing tissue that can evade or withstand attack by the host.75 Our previous reference to a “selective growth advantage” that is reminiscent of Darwinian selection is not accidental. The clonal evolution theory76 addresses the significance of sequential genetic changes providing growth and survival advantages, but to this we must add the fact that, in addition to these self-sufficient events that influence growth and survival, tumor cells must also evade “predators” (e.g., inflammation and the immune system77,78). In essence, the interaction of the tumor with its microenvironment and ultimately with the host is in fact subject to Darwinian laws of evolution, albeit in an accelerated time scale.79 This is evident in the ability of tumors to modulate stromal cells to support their own growth by providing a suitable matrix and an abundance of nutrients, while maintaining antitumor responses at bay. As is true for other selective environments, tumors that outgrow the capability of their immediate surroundings to support their growth must alter that environment to suit their needs or identify other favorable locations where they can become established. The tumor microenvironment was recently shown to exert a significant effect on the complement of genes expressed by incipient tumor cells.80 In this case, the microenvironment was modified by gamma irradiation and the tumor was derived from orthotopic implants of chimeric Trp53-deficient mammary epithelial cells. The magnitude of change was not unlike that observed by our group when comparing the influence of breed on gene expression by canine hemangiosarcoma cells, although in our case the expression profile was maintained in a cell-autonomous fashion (i.e., ex vivo).34 Again, the behavior of carcinomas and sarcomas may differ, and for the latter, recent experiments from our group using canine hemangiosarcoma xenotransplantation models suggest that alterations in the microenvironment might favor not only the efficiency of tumor implantation but also the extent to which the microenvironment contributes to the composition of the tumor as a whole. Incipient sarcoma cells, in turn, can reside as quiescent inhabitants of distant microenvironments themselves modulating growth, morphology, and behavior of microenvironment constituents in the process of metastatic dissemination (Figure 1-6). The concept of genomic instability is not new but was incorporated as an “enabling hallmark” into the updated Hanahan and Weinberg model. Step-wise clonal evolution is satisfying because it can be correlated with discrete pathologic changes in tumor progression, especially for epithelial tumors where such progression can be seen in lesions that go through stages of hyperplasia, atypical hyperplasia (dysplasia), adenoma, carcinoma in situ, invasive carcinoma, and metastatic carcinoma (Figure 1-7). However, analysis of tumor genomes even in early stages usually shows aneuploidy (abnormal DNA copy number), as well as chaotic changes indicative of multiple numeric and structural DNA abnormalities. Similar abnormalities first noticed by Boveri more than 100 years ago in studies of sea urchin cells led him to formulate the “aneuploidy theory” of cancer.81 We know now that aneuploidy is especially evident in solid tumors; based on this, Loeb proposed the existence of the “mutator phenotype” in which cells are predisposed to undergo multiple mutations, some of which inevitably lead to cancer (see earlier).26 Some tenets of his hypothesis appear to be correct, although perhaps in different circumstances than Loeb originally envisioned, because they might relate to increased activity of polymerases with low fidelity under conditions in which the rate of DNA damage (and consequently mutations) is higher than the expected background from normal DNA replication (e.g., in lung epithelial cells from heavy smokers). However, direct measurements of mutation rates of sporadic tumors are much lower than those predicted if a “mutator phenotype” was operative in these tumors.12 Indeed, the minimum number of mutations that are required for clinical onset of cancer in solid tumors based on sequencing of solid tumor genomes is 15 to 25,4 but this may apply to tumors with chaotic karyotypes, as the number of mutations identified in a cytogenetically stable leukemia was significantly smaller.3 Still, genetic instability is a hallmark of most tumors, and while it can be partly explained by increased errors in DNA replication and chromosomal segregation in cells that are rapidly dividing, other mechanisms are clearly operative, involving telomeres and telomerase.12,79,82–85 Although many of these changes are not “recurrent” and appear to be random products of instability, some may in fact contribute to proliferative crisis.86 It is possible that the initiation events for many tumors occur early in life during highly proliferative stages of tissue growth and remodeling (e.g., prior to closure of the growth plates in bone cancer), but they become evident later in life when a last series of mutations allows the transformed cell to reach this crisis stage. As we alluded earlier, hematopoietic tumors seem to avoid the chaotic chromosomal instability associated with solid tumors. We do not fully understand the reasons for this, although it may be partly due to intrinsic protective mechanisms associated with the proliferative rate of bone marrow precursor cells. The role of inflammation in cancer has received considerable attention in the past 10 years. Although our understanding of this phenomenon remains incomplete, it clearly met the criteria for inclusion as an “enabling hallmark” into the updated Hanahan and Weinberg model. The importance of inflammation was inferred from the earliest microscopic studies of cancer, but it was a seminal paper by Dvorak in 1986, in which he described tumors as “wounds that never heal,”87 that provided synthesis for the recurrent observation that tumors were often infiltrated by inflammatory cells of the innate (granulocytes, histiocytes, and macrophages) and the adaptive (lymphocytes) immune systems. Mechanistic distinctions remain to be defined between inflammation that favors tumor growth and inflammation that retards growth or eliminates the tumor,88–90 but we can say confidently that inflammation contributes to tumor growth and survival by supplying factors that sustain proliferation; factors that limit cell death; proangiogenic factors; extracellular matrix-modifying enzymes that facilitate angiogenesis, invasion, and metastasis; and other signals that lead to activation of EMT and other hallmark-facilitating programs.11 As noted earlier, inflammatory cells also release notably reactive oxygen species that are actively mutagenic for nearby cancer cells, accelerating their genetic evolution toward states of heightened malignancy.91 It is worth noting that this characteristic of tumor cells provides at least one important diagnostic advantage. Upregulation of the major glucose transporter, GLUT-1, is seen in virtually all tumors, making the cells efficient glucose scavengers. This can be exploited to image tumor cells noninvasively with precision by visualizing glucose uptake using positron-emission tomography (PET) with a radiolabeled analog of glucose (18F-fluorodeoxyglucose [18F-FDG]) as a reporter. The combination of PET with computed tomography (PET-CT) is now one of the most robust means to evaluate composition of tumors, minimal residual disease, and tumor-specific objective responses in patients receiving conventional and experimental therapies. Burnet and Thomas proposed the concept that the immune system can recognize and destroy incipient tumors (cancer immunosurveillance) in the 1950s.78 However, the hypothesis was far ahead of its time, and technologic obstacles impeded proof, so the theory fell into disfavor. In recent years, the immunosurveillance theory has gained traction anew because data strongly suggest that the immune system helps to maintain tumors at bay, and thus tumors must evade the immune response to survive. In its recent incarnation, the theory has been refined to incorporate the concept of immunoediting, in which the immune system destroys strongly antigenic tumor cells, providing weakly antigenic cells a survival advantage.78 Experimental evidence for this concept includes differences between tumors grown in immunocompetent (only weakly antigenic tumors survive) and immunocompromised mice (no selection against strongly antigenic tumors is observed), but it is unknown if immunoediting is operative in spontaneous cancers. That the tumor microenvironment forms and maintains an immunosuppressive barrier provides more compelling evidence for the role of the immune system to limit tumor growth and metastasis. This immunosuppressive barrier includes cellular factors such as regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), and MSCs. Tregs, MDSCs, and MSCs can attack distinct and complementary antigen-specific (Tregs) and nonspecific (MDSCs and MSCs) facets of immune effector cell activation and function. Soluble factors, including transforming growth factor-β (TGF-β) and immunoglobulins, also contribute to the immunosuppressive barrier directly by disabling immune effector cells and indirectly by “educating” tumor-associated stromal cells, which in turn promotes secretion of stromal-derived factors that recruit additional inflammatory cells (tumor macrophages) and endothelial cells that further accelerate tumor growth.92 This is an active area of basic and clinical research in which companion animal oncology has been at the forefront, for example, through the generation and approval of the first active gene-based therapeutic cancer vaccine for canine melanoma.93 Another observation is that events leading to cancer need not necessarily be caused by mutational events but instead can be caused by epigenetic changes. Epigenetic events are those that can alter phenotype without changing the genotype. Two well-characterized epigenetic mechanisms regulate gene expression. Gene silencing can occur by methylation of CpG residues in promoter regions, as well as by histone deacetylation. Both of these events interfere with the transcriptional machinery and repress gene expression. The effects of global changes in methylation or deacetylation (e.g., by inactivation of DNA methylases or histone deacetylases) remain incompletely understood, but silencing of specific genes by methylation is implicated in numerous cancers of humans and animals.2,94–96 One important observation is that most (or all) genes that are subject to silencing by methylation in specific cancers (e.g., CDKN2A in T-cell leukemia) are commonly inactivated by mutation or deletion in other cancers (e.g., CDKN2A in melanoma). As is true for mutations, gene regulation by epigenetic methylation can occur sporadically or it can be heritable. Silencing of some tumor suppressor genes in sporadic cancers occurs more frequently by epigenetic methylation than by mutation or deletion. These different mechanisms of gene silencing are not equivalent, as they each result in specific tumor phenotypes. For example, data from our laboratories indicate that loss of canine chromosome 11, with resultant deletion of the INK4 tumor suppressor locus containing the CDKN2A, CDKN2B, and ARF genes, and methylation of CDKN2A are each associated with morphologically distinct types of T-cell lymphoma that have a different clinical presentation and prognosis.37,51 Genomic imprinting presents a unique example in which heritable epigenetic changes influence cancer predisposition. Genomic imprinting refers to a pattern of gene expression that is determined by the parental origin of the gene; in other words, unlike most genes in which both parental alleles are expressed, only one allele (specifically derived from the mother or from the father, depending on the gene) of an imprinted gene is expressed and the other one is permanently repressed. Epigenetic changes in Wilms’ tumor and in heritable colon cancer (among others) alter the expression of the imprinted allele, leading to loss of imprinting that causes overexpression of the insulin growth factor-2 (IGF2) gene.2,97 The paradoxic nature of some cancers gave rise to the notion of a “cancer progenitor” or a CSC as far back as the 1960s. The best illustration for this concept was CML, where the bulk of the tumor consists of terminally differentiated neutrophils that are incapable of recreating the malignancy. However, it was apparent that there were multipotent stem cells in this tumor population. In 1994, Dick’s group proved conclusively that another type of leukemia, acute myelogenous leukemia (AML), was a hierarchically organized disease in which a small number of cells that were undetectable by conventional methods could be isolated from patients and made to recapitulate the full spectrum of the disease in an animal model.98 This gave rise to the CSC or “tumor-initiating cell” hypothesis, which is based on the concept that tumors are hierarchically organized into a subpopulation of cells that retain or acquire the capacity for self renewal and are capable and responsible for initiating and maintaining the tumor (Figure 1-8).99 Another subpopulation of cells that consists of the CSC progeny undergo partial to complete differentiation and lose the capability to support the tumor, albeit they still contribute to the morbidity of cancer. This hypothesis fundamentally altered the way we understand cancer but also gave rise to a debate regarding how widely this model applies. The competing hypothesis is based on a model where all the cells in a tumor possess an equal capacity for self-renewal and is commonly referred to as the stochastic model. According to this model, the process of cancer is driven entirely (or almost entirely) by environmental selection of favorable mutations; this model would necessarily predict that cancer is an inevitable outcome for multicellular organisms, and few, if any, long-lived animals would reach reproductive age.100 Thus this model must, by necessity, invoke the existence of protective mechanisms that are independent of cancer risk (e.g., efficient DNA repair mechanisms and immune surveillance). • Figure 1-8 Multiple facets to cancer stem cell (CSC) self-renewal. Increasing evidence is emerging to support the notion that CSC self-renewal decisions can be guided by the activation of several pathways, including Wnt, Notch, Hedgehog, and others. A CSC may autonomously trigger the appropriate signaling cascade to maintain self-renewal with minimal niche support. It is likely that some CSCs need the appropriate microenvironment to provide the stimuli for uncontrolled self-renewal. Finally, some cancer cells have lost the capacity to self-renew regardless of stimulating molecules and hence cannot initiate a tumor. Rights were not granted to include this figure in electronic media. Please refer to the printed book. (Reprinted from O’Brien CA, Kreso A, Jamieson CHM: Cancer stem cells and self-renewal, Clin Cancer Res 16(12):3113–3120, 2010, with permission.) Clin Cancer Res It is possible that the two models represent a continuum dependent on the extent to which CSCs undergo asymmetric versus symmetric divisions. Under conditions in which CSC divisions are primarily asymmetric, few CSCs would be apparent and the population would achieve a hierarchical organization, whereas under conditions in which CSCs underwent symmetric divisions, virtually every cell in the tumor would have CSC-like properties and the organization would be more consistent with a stochastic model. The prevailing opinion is that CSCs exist and are characterized both by peculiar phenotypes and defined sets of mutations of a small number of genes.101–103 Other mutations then endow their progeny with limited or extensive capacity to undergo programmed differentiation, thus resulting in the distinct clinical phenotypes that characterize acute and chronic leukemias or high-grade and low-grade solid tumors. The origin of CSCs is among the most important contemporary topics of investigation. CSCs may arise from mutations that occur in bona fide stem cells, they may arise by “de-differentiation” of somatic cells that acquire mutations that endow them with stem cell–like properties, or they may develop by fusion of a transformed cell and a bone marrow–derived stem cell.104 In companion animals, putative CSCs have been identified in hemangiosarcoma, osteosarcoma, brain tumors, and possibly lymphoma.105–108 The genetic basis of cancer is now beyond question. It is estimated that at least five to seven mutational events are required for overt malignant transformation, and genomic instability seems to be necessary to establish a self-renewing population of cells (possibly CSCs) whose progeny expand to cause clinical disease. Ultimately, a subpopulation endowed with metastatic properties that is drug resistant leads to death of the cancer patient. The rate and the flow of information is such that we predict the coming decade will see transformational changes in our perception of how genes interact with the macroenvironment at the organismal level and with the microenvironment in tumors. Although it is possible that cancer in higher vertebrates is an inevitable consequence of evolution,109 improvements in our understanding of fundamental mechanisms that account for malignant transformation and tumor progression will allow us to design strategies to improve quality of life and outcomes for cancer patients. 1. Vogelstein, B, Kinzler, KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. 2. Ponder, BA. Cancer genetics. Nature. 2001;411:336–341. 3. Ley, TJ, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. 4. Sjoblom, T, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. 5. Morin, RD, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476:298–303. 6. Pasqualucci, L, et al. Analysis of the coding genome of diffuse large B-cell lymphoma. Nat Genet. 2011;43:830–837. 7. Fearon, ER. Human cancer syndromes: clues to the origin and nature of cancer. Science. 1997;278:1043–1050. 8. Nagy, R, Sweet, K, Eng, C. Highly penetrant hereditary cancer syndromes. Oncogene. 2004;23:6445–6470. 9. Knudson, AG. Mutation and cancer: Statistical study of retinoblastoma. Proc Natl Acad Sci U S A. 1971;68:820–823. 10. Hahn, WC, Weinberg, RA. Modelling the molecular circuitry of cancer. Nat Rev Cancer. 2002;2:331–341. 11. Hanahan, D, Weinberg, RA. Hallmarks of cancer: The next generation. Cell. 2011;144:646–674. 12. Pihan, G, Doxsey, SJ. Mutations and aneuploidy: Co-conspirators in cancer? Cancer Cell. 2003;4:89–94. 13. Lingaas, F, et al. A mutation in the canine BHD gene is associated with hereditary multifocal renal cystadenocarcinoma and nodular dermatofibrosis in the German Shepherd dog. Hum Mol Genet. 2003;12:3043–3053. 14. Nickerson, ML, et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt-Hogg-Dube syndrome. Cancer Cell. 2002;2:157–164. 15. Trosko, JE. Commentary: Is the concept of “tumor promotion” a useful paradigm? Mol Carcinog. 2001;30:131–137. 16. Kopelovich, L. Hereditary adenomatosis of the colon and rectum: relevance to cancer promotion and cancer control in humans. Cancer Genet Cytogenet. 1982;5:333–352. 17. Heng, HH. Cancer genome sequencing: the challenges ahead. Bioessays. 2007;29:783–794. 18. Balmain, A. Cancer as a complex genetic trait: tumor susceptibility in humans and mouse models. Cell. 2002;108:145–152. 19. Amos, CI, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet. 2008;40:616–622. 20. Hung, RJ, et al. A susceptibility locus for lung cancer maps to nicotinic acetylcholine receptor subunit genes on 15q25. Nature. 2008;452:633–637. 21. Thorgeirsson, TE, et al. A variant associated with nicotine dependence, lung cancer and peripheral arterial disease. Nature. 2008;452:638–642. 22. Liu, JZ, et al. Meta-analysis and imputation refines the association of 15q25 with smoking quantity. Nat Genet. 2010;42:436–440. 23. Shah, PP, et al. Association of functionally important polymorphisms in cytochrome P4501B1 with lung cancer. Mutat Res. 2008;643:4–10. 24. Hecht, SS, Kassie, F, Hatsukami, DK. Chemoprevention of lung carcinogenesis in addicted smokers and ex-smokers. Nat Rev Cancer. 2009;9:476–488. 25. Jalas, JR, Hecht, SS, Murphy, SE. Cytochrome P450 enzymes as catalysts of metabolism of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco specific carcinogen. Chem Res Toxicol. 2005;18:95–110. 26. Loeb, LA. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res. 1991;51:3075–3079. 27. Lindblad-Toh, K, et al. Breen, M, et al, eds Genes, dogs and cancer: Fifth Canine Cancer Conference. Orlando: IVIS; 2009;vol 5. 28. Ostrander, EA, Breen, M, et al, eds. Genes, dogs and cancer: Fifth Canine Cancer Conference, vol 5. Orlando: IVIS, 2009. 29. Pontius, JU, et al. Initial sequence and comparative analysis of the cat genome. Genome Res. 2007;17:1675–1689. 30. Thomas, R, et al. Microarray-based cytogenetic profiling reveals recurrent and subtype-associated genomic copy number aberrations in feline sarcomas. Chromosome Res. 2009;17:987–1000. 31. Hedan, B, et al. Molecular cytogenetic characterization of canine histiocytic sarcoma: A spontaneous model for human histiocytic cancer identifies deletion of tumor suppressor genes and highlights influence of genetic background on tumor behavior. BMC Cancer. 2011;11:201. 32. Thomas, R, et al. Influence of genetic background on tumor karyotypes: Evidence for breed-associated cytogenetic aberrations in canine appendicular osteosarcoma. Chromosome Res. 2009;17:365–377. 33. Modiano, JF, et al. Distinct B-cell and T-cell lymphoproliferative disease prevalence among dog breeds indicates heritable risk. Cancer Res. 2005;65:5654–5661. 34. Tamburini, BA, et al. Gene expression profiles of sporadic canine hemangiosarcoma are uniquely associated with breed. PLoS ONE. 2009;4:e5549. 35. Scott, MC, et al. Molecular subtypes of osteosarcoma identified by reducing tumor heterogeneity through an interspecies comparative approach. Bone. 2011;49:356–367. 36. Breen, M, Modiano, JF. Evolutionarily conserved cytogenetic changes in hematological malignancies of dogs and humans—man and his best friend share more than companionship. Chromosome Res. 2008;16:145–154. 37. Fosmire, SP, et al. Inactivation of the p16 cyclin-dependent kinase inhibitor in high-grade canine non-Hodgkin’s T-cell lymphoma. Vet Pathol. 2007;44:467–478. 38. Angstadt, AY, et al. Characterization of canine osteosarcoma by array comparative genomic hybridization and RT-qPCR: Signatures of genomic imbalance in canine osteosarcoma parallel the human counterpart. Genes Chromosomes Cancer. 2011;50(11):859–874. 39. Duesberg, P, Li, R. Multistep carcinogenesis: A chain reaction of aneuploidizations. Cell Cycle. 2003;2:202–210. 40. Duesberg, P, Rausch, C, Rasnick, D, et al. Genetic instability of cancer cells is proportional to their degree of aneuploidy. Proc Natl Acad Sci U S A. 1998;95:13692–13697. 41. Mani, RS, Chinnaiyan, AM. Triggers for genomic rearrangements: insights into genomic, cellular and environmental influences. Nat Rev Genet. 2010;11:819–829. 42. O’Donoghue, LE, et al. Expression profiling in canine osteosarcoma: identification of biomarkers and pathways associated with outcome. BMC Cancer. 2010;10:506. 43. Paoloni, M, et al. Canine tumor cross-species genomics uncovers targets linked to osteosarcoma progression. BMC Genomics. 2009;10:625. 44. Selvarajah, GT, et al. Gene expression profiling of canine osteosarcoma reveals genes associated with short and long survival times. Mol Cancer. 2009;8:72. 45. Hanahan, D, Weinberg, RA. The hallmarks of cancer. Cell. 2000;100:57–70. 46. Ruchatz, H, Coluccia, AM, Stano, P, et al. Constitutive activation of Jak2 contributes to proliferation and resistance to apoptosis in NPM/ALK-transformed cells. Exp Hematol. 2003;31:309–315. 47. Modiano, JF, Ritt, MG, Wojcieszyn, J, et al. Growth arrest of melanoma cells is differentially regulated by contact inhibition and serum deprivation. DNA Cell Biol. 1999;18:357–367. 48. Modiano, JF, Johnson, LD, Bellgrau, D. Negative regulators in homeostasis of naive peripheral T cells. Immunol Res. 2008;41:137–153. 49. Levine, RA, Fleischli, MA. Inactivation of p53 and retinoblastoma family pathways in canine osteosarcoma cell lines. Vet Pathol. 2000;37:54–61. 50. Levine, RA, Forest, T, Smith, C. Tumor suppressor PTEN is mutated in canine osteosarcoma cell lines and tumors. Vet Pathol. 2002;39:372–378. 51. Modiano, JF, Breen, M, Valli, VE, et al. Predictive value of p16 or Rb inactivation in a model of naturally occurring canine non-Hodgkin’s lymphoma. Leukemia. 2007;21:184–187. 52. Dickerson, EB, et al. Mutations of phosphatase and tensin homolog deleted from chromosome 10 in canine hemangiosarcoma. Vet Pathol. 2005;42:618–632. 53. Hammerman, PS, Fox, CJ, Thompson, CB. Beginnings of a signal-transduction pathway for bioenergetic control of cell survival. Trends Biochem Sci. 2004;29:586–592. 54. White, E, DiPaola, RS. The double-edged sword of autophagy modulation in cancer. Clin Cancer Res. 2009;15:5308–5316. 55. Ohyashiki, JH, Sashida, G, Tauchi, T, et al. Telomeres and telomerase in hematologic neoplasia. Oncogene. 2002;21:680–687. 56. Pang, LY, Argyle, DJ. Using naturally occurring tumours in dogs and cats to study telomerase and cancer stem cell biology. Biochim Biophys Acta. 2009;1792:380–391. 57. Ferrara, N, Kerbel, RS. Angiogenesis as a therapeutic target. Nature. 2005;438:967–974. 58. Folkman, J. The role of angiogenesis in tumor growth. Semin Cancer Biol. 1992;3:65–71. 59. Folkman, J. Tumor angiogenesis: therapeutic implications. N Engl J Med. 1971;285:1182–1186. 60. Chen, H, et al. In vitro and in vivo production of vascular endothelial growth factor by chronic lymphocytic leukemia cells. Blood. 2000;96:3181–3187. 61. Kini, AR, Kay, NE, Peterson, LC. Increased bone marrow angiogenesis in B cell chronic lymphocytic leukemia. Leukemia. 2000;14:1414–1418. 62. Molica, S, et al. Prognostic value of enhanced bone marrow angiogenesis in early B-cell chronic lymphocytic leukemia. Blood. 2002;100:3344–3351. 63. Juliusson, G, et al. Frequent good partial remissions from thalidomide including best response ever in patients with advanced refractory and relapsed myeloma. Br J Haematol. 2000;109:89–96. 64. Tosi, P, et al. Salvage therapy with thalidomide in multiple myeloma patients relapsing after autologous peripheral blood stem cell transplantation. Haematologica. 2001;86:409–413. 65. Jorgensen, JM. The role of angiogenesis in non-Hodgkin lymphoma. Dan Med Bull. 2005;52:254. 66. Ranieri, G, et al. Endothelial area and microvascular density in a canine non-Hodgkin’s lymphoma: an interspecies model of tumor angiogenesis. Leuk Lymphoma. 2005;46:1639–1643. 67. Preziosi, R, Sarli, G, Paltrinieri, M. Prognostic value of intratumoral vessel density in cutaneous mast cell tumors of the dog. J Comp Pathol. 2004;130:143–151. 68. Restucci, B, et al. Expression of vascular endothelial growth factor receptor Flk-1 in canine mammary tumours. J Comp Pathol. 2004;130:99–104. 69. Fidler, IJ. The pathogenesis of cancer metastasis: the ‘seed and soil’ hypothesis revisited. Nat Rev Cancer. 2003;3:453–458. 70. Kim, JW, et al. Rapid apoptosis in the pulmonary vasculature distinguishes non-metastatic from metastatic melanoma cells. Cancer Lett. 2004;213:203–212. 71. Wong, CW, et al. Intravascular location of breast cancer cells after spontaneous metastasis to the lung. Am J Pathol. 2002;161:749–753. 72. Wong, CW, et al. Apoptosis: an early event in metastatic inefficiency. Cancer Res. 2001;61:333–338. 73. Koshkina, NV, et al. Fas-negative osteosarcoma tumor cells are selected during metastasis to the lungs: the role of the Fas pathway in the metastatic process of osteosarcoma. Mol Cancer Res. 2007;5:991–999. 74. Krishnan, K, et al. Ezrin mediates growth and survival in Ewing’s sarcoma through the AKT/mTOR, but not the MAPK, signaling pathway. Clin Exp Metastasis. 2006;23:227–236. 75. Mueller, MM, Fusenig, NE. Friends or foes—bipolar effects of the tumour stroma in cancer. Nat Rev Cancer. 2004;4:839–849. 76. Nowell, PC. Mechanisms of tumor progression. Cancer Res. 1986;46:2203–2207. 77. Modiano, JF, et al. Fas ligand gene transfer for cancer therapy. Cancer Ther. 2004;2:561–570. 78. Dunn, GP, Bruce, AT, Ikeda, H, et al. Cancer immunoediting: from immunosurveillance to tumor escape. Nat Immunol. 2002;3:991–998. 79. Breivik, J. The evolutionary origin of genetic instability in cancer development. Semin Cancer Biol. 2005;15:51–60. 80. Nguyen, DH, et al. Radiation acts on the microenvironment to affect breast carcinogenesis by distinct mechanisms that decrease cancer latency and affect tumor type. Cancer Cell. 2011;19:640–651. 81. Boveri, T. Concerning the origin of malignant tumors. J Cell Sci. 2008;121:1–84. 82. Albertson, DG, Collins, C, McCormick, F, et al. Chromosome aberrations in solid tumors. Nat Genet. 2003;34:369–376. 83. Teixeira, MR, Heim, S. Multiple numerical chromosome aberrations in cancer: what are their causes and what are their consequences? Semin Cancer Biol. 2005;15:3–12. 84. Gollin, SM. Mechanisms leading to chromosomal instability. Semin Cancer Biol. 2005;15:33–42. 85. Rajagopalan, H, Lengauer, C. Aneuploidy and cancer. Nature. 2004;432:338–341. 86. Maser, RS, DePinho, RA. Connecting chromosomes, crisis, and cancer. Science. 2002;297:565–569. 87. Dvorak, HF. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med. 1986;315:1650–1659. 88. Lin, WW, Karin, M. A cytokine-mediated link between innate immunity, inflammation, and cancer. J Clin Invest. 2007;117:1175–1183. 89. Mantovani, A, Allavena, P, Sica, A, et al. Cancer-related inflammation. Nature. 2008;454:436–444. 90. Bhatia, R, McGlave, PB, Dewald, GW, et al. Abnormal function of the bone marrow microenvironment in chronic myelogenous leukemia: role of malignant stromal macrophages. Blood. 1995;85:3636–3645. 91. Grivennikov, SI, Greten, FR, Karin, M. Immunity, inflammation, and cancer. Cell. 2010;140:883–899. 92. Erez, N, Truitt, M, Olson, P, et al. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NF-kappaB-dependent manner. Cancer Cell. 2010;17:135–147. 93. Bergman, PJ. Anticancer vaccines. Vet Clin North Am Small Anim Pract. 2007;37:1111–1119. [vi-ii]. 94. Wolffe, AP, Matzke, MA. Epigenetics: regulation through repression. Science. 1999;286:481–486. 95. Costello, JF. Comparative epigenomics of leukemia. Nat Genet. 2005;37:211–212. 96. Yu, L, et al. Global assessment of promoter methylation in a mouse model of cancer identifies ID4 as a putative tumor-suppressor gene in human leukemia. Nat Genet. 2005;37:265–274. 97. Cui, H, et al. Loss of IGF2 imprinting: a potential marker of colorectal cancer risk. Science. 2003;299:1753–1755. 98. Lapidot, T, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. 99. O’Brien, CA, Kreso, A, Jamieson, CHM. Cancer stem cells and self-renewal. Clin Cancer Res. 2010;16(12):3113–3120. 100. Clarke, MF, Fuller, M. Stem cells and cancer: two faces of eve. Cell. 2006;124:1111–1115. 101. Huntly, BJ, Gilliland, DG. Leukaemia stem cells and the evolution of cancer-stem-cell research. Nat Rev Cancer. 2005;5:311–321. 102. Singh, SK, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. 103. Smith, GH. Mammary cancer and epithelial stem cells: a problem or a solution? Breast Cancer Res. 2002;4:47–50. 104. Bjerkvig, R, Tysnes, BB, Aboody, KS, et al. Opinion: the origin of the cancer stem cell: current controversies and new insights. Nat Rev Cancer. 2005;5:899–904. 105. Lamerato-Kozicki, AR, Helm, KM, Jubala, CM, et al. Canine hemangiosarcoma originates from hematopoietic precursors with potential for endothelial differentiation. Exp Hematol. 2006;34:870–878. 106. Wilson, H, et al. Isolation and characterisation of cancer stem cells from canine osteosarcoma. Vet J. 2007;175(1):69–75. 107. Stoica, G, et al. Identification of cancer stem cells in dog glioblastoma. Vet Pathol. 2009;46(3):391–406. 108. Ito, D, et al. A tumor-related lymphoid progenitor population supports hierarchical tumor organization in canine B-cell lymphoma. J Vet Intern Med. 2011;25:890–896. 109. Modiano, JF, Breen, M. Shared pathogenesis of human and canine tumors—an inextricable link between cancer and evolution. Cancer Ther. 2008;6:239–246.
The Etiology of Cancer
 Section A
Section A
The Genetic Basis of Cancer
Heritable Cancer Syndromes
Genetic Influence in Sporadic Cancers
The Hallmarks of Cancer
Self-Sufficiency of Growth Signals
Insensitivity to Antigrowth Signals
Evasion of Cell Death
Limitless Replicative Potential
Sustained Angiogenesis
Invasion and Metastasis
Adaptive Evolution and the Tumor Microenvironment
Emerging and Enabling Hallmarks
Tumor-Promoting Inflammation
Reprogramming Energy Metabolism
Evading Immune Destruction
Epigenetic Events
Cancer Stem Cells

Summary
References
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


The Etiology of Cancer