and Paul Vezina1
(1)
Department of Psychiatry and Behavioral Neuroscience, The University of Chicago, Chicago, IL, USA
Abstract
Exposure to psychostimulant drugs leads to sensitized locomotor responding, nucleus accumbens (NAcc) dopamine (DA) overflow, and drug self-administration upon reexposure to the drug weeks to months later. Calcium-dependent signaling pathways contribute importantly to both the induction and expression of sensitization by these drugs. In particular, calcium-calmodulin (CaM)-dependent protein kinase II (CaMKII), a serine/threonine kinase that is highly expressed in forebrain regions such as the NAcc, is known to contribute to the expression of several forms of plasticity including sensitization. Notably, pharmacologically inhibiting CaMKII in the NAcc prevents the expression of sensitized locomotion, NAcc DA overflow, and drug taking. Evidence indicates that CaMKII may act both pre- and postsynaptically in this site to influence the expression of these manifestations of sensitization. Presynaptically in DA neuron terminals, CaMKII regulates sensitized DA overflow. Postsynaptically in medium spiny neurons, it contributes to the upregulation of AMPA receptors, the activation of which is necessary for the expression of sensitization. It is conceivable that interactions between CaMKII-mediated neuroadaptations in both of these sites may underlie the long-lasting maintenance of sensitization by psychostimulant drugs.
Key words
CaMKIIDopamineDrug self-administrationGlutamateLocomotionNucleus accumbensReinstatementSensitization1 Stimulant Sensitization
Psychostimulant drugs like amphetamine and cocaine increase extracellular levels of dopamine (DA) in brain. They achieve this effect in the somatodendritic and terminal regions of DA neurons by blocking uptake of DA into the cell or causing reverse transport of DA through the DA transporter into the extracellular space (1). Several lines of evidence indicate that the locomotor-activating, reinforcing, and incentive effects of psychostimulants are mediated by transmission in the mesocorticolimbic DA pathways. These pathways consist of DA cell bodies located in the midbrain ventral tegmental area (VTA) and their ascending projections primarily to the nucleus accumbens (NAcc) as well as other brain regions including the prefrontal cortex, hippocampus, and amygdala (2). It has been known for some time that repeated exposure to psychostimulants leads to sensitized locomotor responding to these drugs when rats are reexposed to the drug weeks to months later (3). This long-lasting effect is paralleled by equally long-lasting enhancements in NAcc DA neuron reactivity (4–6). More recently, it was shown that repeated exposure to psychostimulants also facilitates acquisition of drug self-administration (7, 8) and enhances the subsequent reinstatement of drug seeking (9) as well as the amount of work animals will emit to self-administer the drug (10). Evidence indicates that sensitization of stimulant-induced locomotion, drug self-administration, and NAcc DA overflow are produced by the same drug exposure regimens via actions in the same nuclei (5, 10, 11) and are prevented by the same pharmacological interventions (6, 12–14). Such findings provide strong support for the idea that the enhanced expression of complex motivated behaviors like drug seeking and drug taking represent, like enhanced locomotion, important manifestations of behavioral sensitization. Thus, sensitization of midbrain DA neuron reactivity has been proposed to underlie the transition from casual drug use to craving and abuse (15, 16). The growing number of findings showing that enhanced behavioral and neuronal responding to stimulants also occurs in humans (see (17, 18)) highlights the importance of understanding sensitization at the molecular, cellular, systems, and behavioral levels so as to promote the development of the therapeutic interventions necessary to reverse it.
Activity in VTA-NAcc DA neurons has long been associated, not only with drug-induced locomotion, but also with self-administration supported by amphetamine and other psychostimulant drugs. For example, both effects are prevented by DA receptor blockade or 6-OHDA lesions of DA neuron terminals in the NAcc (for discussion and references, see (16)). It would thus be expected that the progressive enhancement in the reactivity of these neurons should lead to the progressive enhancement of the manifestation of both of these behavioral outputs, as the above results indicate. In their seminal paper (15), Robinson and Berridge proposed that activity in VTA-NAcc DA neurons encodes the incentive valence of drug cues. Sensitization of the reactivity of these neurons would thus be expected to enhance the incentive valence of these cues, allowing them to more readily illicit approach and as a result to augment the pursuit of the drug, a result again indicated by the above findings.
Stimulant sensitization can be divided into two distinct components, each associated with a separate brain region and different sets of identified neuroadaptations. Induction takes place in midbrain nuclei such as the VTA and expression occurs in forebrain sites like the NAcc (19, 20). The former encompasses the transient neuronal events that are produced by repeated, intermittent exposure to the drug. Because the VTA is richly innervated, a number of neurotransmitter systems have been implicated in the induction of sensitization with glutamate and somatodendritically derived DA proposed to play a major role in this effect (16, 21). Pharmacological activation and inhibition studies suggest an important role in this site for calcium-initiated signaling pathways including those involving calcium-calmodulin (CaM)-dependent protein kinase II (CaMKII) and mitogen-activated protein kinase, as well as protein kinase A and C (16, 22). As a site of expression, the NAcc is home to a number of long-lasting alterations in neuronal signaling that have been linked to enhanced neurochemical and behavioral responding to psychostimulant drugs. Of all neuroadaptations known to follow exposure to such drugs, the one most consistently associated with the expression of locomotor sensitization is the enhancement in the ability of psychostimulants to increase extracellular levels of DA in the NAcc (5, 6, 16). A number of other long-lasting adaptations have also been reported to occur in this site, including alterations in dendritic morphology (23), increased glutamate overflow (24, 25), functional upregulation of AMPA receptors (9, 25), changes in synaptic strength at cortico-accumbens glutamate synapses (26), as well as changes in AMPA receptor subunit expression and membrane insertion (27–29). As with induction in the VTA, CaMKII is again one of a number of proteins implicated in the expression of stimulant sensitization (30–33). Others include PKA, Darpp-32, cAMP response element binding protein, extracellular signal-regulated kinase, and immediate early gene Fos-related antigens like delta FosB (34–36). Of these, CaMKII has attracted considerable attention for its potential role in long-term memory storage due to its abundance in brain, its prominence in the postsynaptic density and its ability to remain phosphorylated after activation (37). CaMKII is also well positioned in post-receptor signaling pathways to bridge converging DA and glutamate neurotransmission in the NAcc (38). Considering that stimulant sensitization is long lasting and that both DA and glutamate contribute to its expression, CaMKII has been investigated particularly for its contribution to the expression of sensitization by psychostimulant drugs. The evidence for this contribution is reviewed below.
2 CaMKII Structure and Function
CaMKII is a serine/threonine kinase that belongs to the multifunctional CaMK family (CaMKI, II, and IV) and phosphorylates a wide array of downstream targets. CaMKII is highly expressed in forebrain sites like the NAcc, hippocampus, amygdala, and prefrontal cortex (39, 40). Four different CaMKII subunits have been identified: α, β, γ, and δ. The α and β subunits are the principal isoforms found in brain, where they form dodecameric holoenzymes made up of one or both subunits (41, 42). In rat forebrain, the α subunit predominates at a ratio of approximately 3:1 over the β subunit (41, 43).
The unique activation properties of this kinase play an important role in its ability to significantly alter synaptic strength and neuronal activity, and thus, it has attracted attention as a major contributor to various types of neuronal plasticity including the sensitization produced by psychostimulant drugs. When CaM is not bound to the kinase, the autoinhibitory domain binds to and occludes the catalytic domain, where the substrate-binding site is located (Fig. 1). With rising calcium levels, CaM binds to the autoinhibitory domain and temporarily activates the kinase (44). This sequence of events also exposes a threonine residue (Thr286) on the autoinhibitory domain and permits its autophosphorylation, thereby preventing inactivation of CaMKII. In this constitutive active state, CaMKII can remain active in the absence of CaM and thus exhibit calcium-independent activity (42, 45). This property of CaMKII has been proposed to contribute importantly to its impact on neuronal activity and especially to its role in the production of long-term potentiation, a primary model of the cellular and molecular mechanisms underlying learning and memory (37). It remains unknown whether this constitutive activity of CaMKII is also necessary either for the induction or the long-term maintenance of stimulant sensitization. Considering its wide array of substrates, it is equally feasible that CaMKII acts as a trigger to initiate downstream post-phosphorylation cascades that themselves mediate the long-lasting expression of sensitized responding to psychostimulant drugs. Whichever the case, a number of reports have indicated that CaMKII is in fact necessary for the expression of stimulant sensitization.
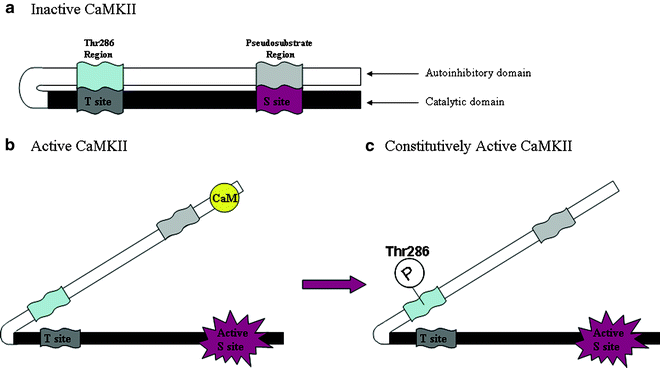
Fig. 1.
Illustration of the principal functional domains of CaMKII. When CaMKII is inactive (a), the Thr286 (α-isoform; Thr287 on the β-isoform) and pseudosubstrate segments of the autoinhibitory domain bind to the T site and substrate-binding site (S site) of the catalytic domain, respectively, to prevent kinase activity. Binding of CaM to a region overlapping with the pseudosubstrate region removes it and the autoinhibitory domain from the S site and activates the kinase (b). Once the kinase is active and the autoinhibitory domain has been removed from the catalytic domain, the latter can now be autophosphorylated at Thr286 (c), thereby preventing binding of this segment to the T site and deactivation of the kinase. Binding of the T site to a region on the NMDA NR2B subunit produces the same effect. This constitutively active form of CaMKII no longer requires CaM to remain active, exhibits calcium-independent activity, and has been proposed to serve as a molecular switch capable of long-term memory storage (37).
3 CaMKII and the Expression of Stimulant Sensitization: Behavioral Evidence
As indicated above, CaMKII has been shown to be necessary for the induction of long-term potentiation (37, 46) and locomotor sensitization by psychostimulant drugs (47). Although there have been relatively few investigations, the evidence obtained to date indicates that CaMKII is also necessary for the expression of behavioral sensitization by cocaine and amphetamine (Fig. 2). For example, microinjecting the CaMKII inhibitor KN-93 into the shell of the NAcc attenuates the expression of locomotor sensitization in rats previously exposed to cocaine. Similar findings were obtained following infusions into the NAcc of L-type calcium channel blockers (31, 48). More recently, infusion of KN-93 into the NAcc shell, but not into the NAcc core, was shown to decrease the enhanced work output and self-administration of amphetamine normally observed in rats previously exposed to the drug (33). These findings indicate that CaMKII is necessary for the enhanced expression of locomotor activity as well as complex motivated behaviors, both behavioral correlates of sensitized midbrain DA neurotransmission (16). Indeed, several reports indicate that CaMKII is necessary for the expression of the latter as well.
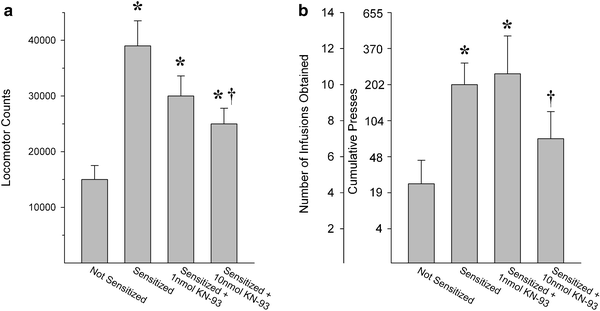
Fig. 2.
Inhibition of CaMKII in the NAcc shell decreases the enhanced locomotion and drug self-administration normally observed in rats previously exposed to psychostimulant drugs. In both cases, the effect of increasing concentrations of the CaMKII inhibitor KN-93 on the expression of sensitized behavior was assessed. Data are shown as (a) 2-h total locomotor activity counts (group means+SEM) following a systemic injection of cocaine or (b) amphetamine infusions (group means+SEM) obtained on a progressive ratio schedule of reinforcement (cumulative presses required are also shown). KN-93 was administered before the cocaine injection in (a) and before the amphetamine self-administration session in (b). Non-sensitized behaviors were not affected by KN-93 (not shown). Significantly different from: * the not-sensitized condition; † the sensitized condition (Adapted from (31, 33). With kind permission).
4 CaMKII: Presynaptic Effects
Amphetamine is known to acutely increase extracellular DA levels via a non-calcium-dependent mechanism. However, the enhanced portion of DA overflow observed in the NAcc of rats previously exposed to amphetamine does require calcium. For example, L-type (diltiazem) and N-type (conotoxin) calcium channel blockers inhibit the expression of sensitized NAcc DA overflow by amphetamine while sparing the ability of the drug to increase overflow acutely (30). These results obtained in vivo do not rule out the recruitment by calcium of postsynaptic signaling pathways in medium spiny neurons and their modulation of DA neuron firing via descending projections to the VTA. Nonetheless, it is likely that the entry of calcium into DA neuron terminals in the NAcc modulates, at least in part, the expression of amphetamine sensitization and that it does so by activating CaMKII. Inhibiting this kinase in the NAcc blocks the expression of sensitized DA overflow in this site (30). Reports showing that application of the CaMKII inhibitor KN-93 to striatal slices from amphetamine-exposed rats blocks sensitized DA release support a presynaptic site of action ((32), Fig. 3). These findings indicate that CaMKII can enhance DA release in the striatum, and by extension in the NAcc, through a presynaptic mechanism by interacting with substrates within the terminals of midbrain DA neurons. Although CaMKII is known to phosphorylate synapsin I at a site that frees synaptic vesicles for exocytosis (49), this kinase appears to mediate enhanced amphetamine-induced DA release via a non-exocytotic mechanism. Inhibition of CaMKII, for example, does not block enhanced K+-evoked DA release in rat striatal slices and maintains its ability to block enhanced amphetamine-induced DA release even after depletion of DA vesicles by reserpine (32). CaMKII may thus mediate enhanced amphetamine-induced NAcc DA overflow by phosphorylating the dopamine transporter or proteins in contact with it at several potential consensus sites (50). For example, there is evidence that CaMKII promotes acute amphetamine-induced DA release by interacting with the C-terminus of the DA transporter (51) or by supporting association of the transporter with syntaxin 1A (52). Alterations in these or other interactions could conceivably lead to enhanced DA release. While the mechanisms whereby CaMKII promotes the expression of sensitized NAcc DA release remain elusive, taken together these findings suggest that calcium-mediated second messenger pathways are altered in sensitized midbrain DA neurons and are well positioned to mediate enhanced DA release. Given that drug self-administration is enhanced in animals sensitized to amphetamine and is accompanied by enhanced reactivity in midbrain DA neurons, these presynaptic calcium signaling pathways may be important for the expression of excessive drug taking.
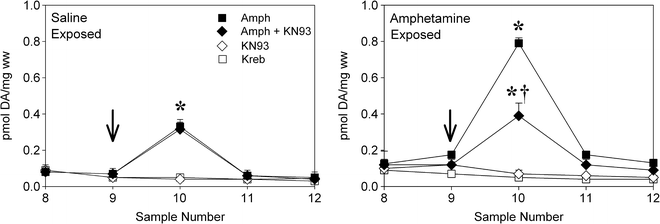
Fig. 3.
Inhibition of CaMKII prevents the enhanced amphetamine-induced DA release normally observed in striatal slices obtained from rats previously exposed to amphetamine (b). Slices prepared from rats previously exposed to saline are shown in (a). Slices were perfused with Kreb buffer and amphetamine (Amph), the CaMKII inhibitor KN-93, or a combination of the two was introduced for 2.5 min starting at the arrow. Samples were collected every 5 min. Data are shown as group mean (±SEM) pmol DA released per mg wet weight tissue per sample. Significantly different from: *, the KN-93 and Kreb-alone conditions; †, the amphetamine-alone condition (Adapted from (32). With kind permission).
Levels of calmodulin (which binds calcium to form CaM that activates CaMKII) are elevated following exposure to amphetamine. This has been demonstrated in striatal tissue (53–55) and striatal synaptosomes (56). As CaM activates CaMKII, changes in calmodulin protein levels following exposure to amphetamine would be expected to alter CaMKII activity. Indeed, CaMKII activity (but not protein levels) has been reported to be increased in striatal synaptosomes prepared from amphetamine-exposed rats (49), again supporting a presynaptic site of action for CaMKII in sensitized amphetamine-induced DA release. Interestingly, findings obtained in the NAcc have been less consistent. In this site, previous exposure to amphetamine has been reported to decrease calmodulin levels (57) and to leave levels of CaMKII and its constitutively active form unchanged (58). Thus, it is possible that CaMKII contributes differentially to the expression of sensitization in the striatum and the NAcc. Alternatively, as both of these reports assayed these proteins in NAcc punches or hand-dissected tissues as opposed to synaptosomes, it is conceivable that the results obtained were influenced by different alterations in CaMKII signaling in pre- and postsynaptic sites (59). As with its presynaptic actions, CaM may also contribute to the initiation of neuroadaptations by activating CaMKII postsynaptically where it recognizes numerous substrates.
5 CaMKII: Postsynaptic Effects
CaMKII is ubiquitously expressed in brain and is abundant in both pre- and postsynaptic sites where it phosphorylates many functional proteins (60). The GluR1 subunit of AMPA receptors expressed by intrinsic NAcc neurons is known to express such targets. CaMKII directly phosphorylates the AMPA receptor GluR1 subunit at serine residue 831 to increase unitary conductance (61, 62) and response quantal size (63). In addition, GluR1-containing receptors are delivered to synapses in an activity-dependent manner that involves entry into the extrasynaptic site and lateral movement into the synapse (64). CaMKII activity is also required for insertion of GluR1 into the cell membrane although the specifically targeted substrate remains unknown (65). These CaMKII-mediated, activity-dependent changes in AMPA receptor function are known to play an important role in altering synaptic strength and producing long-term potentiation (66, 67). It is likely that they also underlie sensitized behavioral responding to psychostimulant drugs as changes in glutamatergic transmission are known to contribute importantly to the expression of stimulant sensitization (21). For example, AMPA receptor antagonists, administered either systemically or into the NAcc, block the expression of locomotor sensitization by AMPH or cocaine (68–71); cf (72). Conversely, microinjection of AMPA into the NAcc produces enhanced cocaine-induced locomotor activity (25, 73) and reinstatement of cocaine seeking (9) in stimulant exposed rats compared to nonexposed controls. Rats showing sensitized locomotor responding to cocaine also display enhanced levels of GluR1 surface expression (27, 28, 74) and increased AMPA/NMDA receptor ratios in the NAcc (26), both findings indicative of enhanced synaptic excitability in this site. Finally, increased membrane insertion of AMPA receptors lacking the GluR2 subunit have been shown to mediate enhanced cue-induced cocaine seeking (29). In these cross-linking experiments, AMPA receptors detected on the cell surface were found to be associated with the postsynaptic protein PSD95 (27). Notably, experiments conducted in co-cultures of primary NAcc and prefrontal cortex neurons showed that CaMKII activity is required for the enhanced cell surface expression of AMPA receptors in NAcc neurons following repeated DA (75). Taken together, these findings are consistent with the possibility that CaMKII activity in intrinsic NAcc neurons regulates postsynaptic glutamate-dependent mechanisms underlying the expression of sensitization.
Evidence, more directly illustrating a role for CaMKII in NAcc neurons in the expression of behavioral sensitization by amphetamine, was recently obtained in experiments using viral-mediated gene transfer. In these experiments, herpes simplex viral (HSV) vectors were microinjected into the NAcc to overexpress CaMKII in neurons adjacent to the injection site (76). Using vectors expressing green florescent protein, whole brain immunohistochemical analyses revealed that infection was limited to these neurons in the NAcc and that neurons in nuclei sending projections to this site were spared (77). Remarkably, overexpressing CaMKII in the NAcc of drug naïve rats produced a sensitized locomotor response to amphetamine (78) and increased work output and amphetamine self-administration (79) relative to control rats that were not infected or infected with a reporter gene. As illustrated in Fig. 4, infected rats showed a sensitized locomotor response to a challenge injection of amphetamine even though they had not previously been exposed to the drug. These findings extend those described above and strongly support an important role for CaMKII signaling in NAcc neurons in the expression of stimulant sensitization. Thus, overexpressing CaMKII in the NAcc may mimic some of the changes produced by amphetamine exposure that lead to long-term maintenance of sensitization. Together with the findings reviewed earlier, these results also suggest that overexpressing CaMKII may enhance behavioral responding to amphetamine by functionally upregulating AMPA receptors in these neurons.
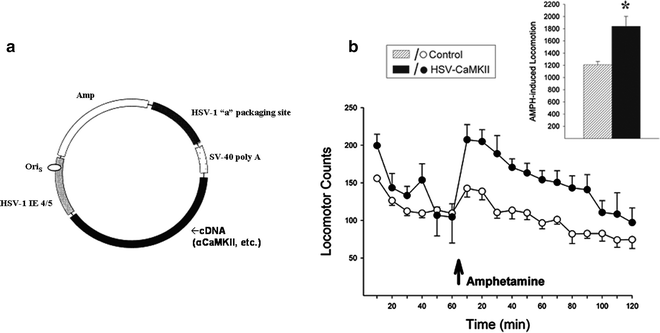




Fig. 4.
Overexpressing CaMKII in the NAcc of drug-naïve rats enhances locomotor responding to amphetamine. Herpes simplex virus vectors were constructed by inserting rat CaMKII cDNA into the vector pHSV-PrpUC, depicted in (a). The main elements of the vector necessary for packaging and transgene expression are the packaging site (HSV-1 “a”), the immediate early gene promoter (HSV-1 IE 4/5), the sequence of origin (OriS), and the simian virus 40 (SV-40) polyadenylation (poly A) signal. The transgene was subcloned into the HSV amplicon, transfected into 2–2 African green monkey kidney epithelium cells, and superinfected with replication-deficient helper virus. After several passages, the virus was purified, pelleted, and resuspended in 10% sucrose. (b) Preliminary data demonstrating that viral-mediated overexpression of CaMKII in the NAcc of drug-naïve rats leads to enhanced locomotor responding to amphetamine when compared to control rats that were not infected (10% sucrose infusions) or infected with a reporter gene (HSV-LacZ). Data are shown as locomotor activity counts (group means±SEM) obtained before and after a systemic injection of amphetamine (arrow). Inset shows 2-h total locomotor activity counts obtained after amphetamine. *Significantly different from controls (78) (a: Adapted from (88). With kind permission).
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
