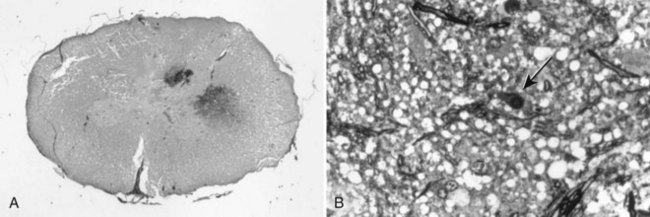
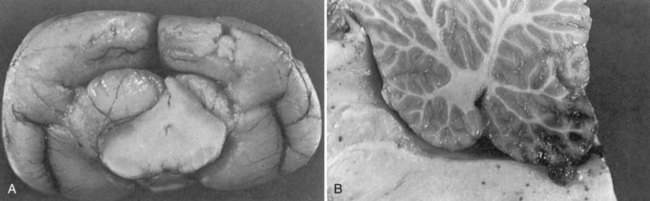
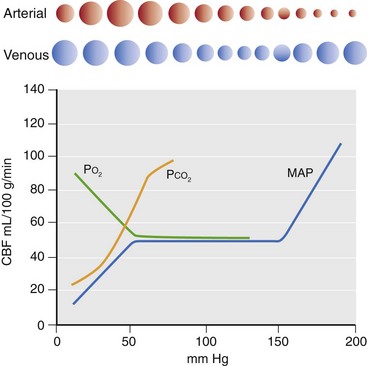
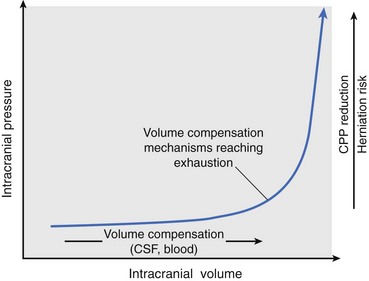
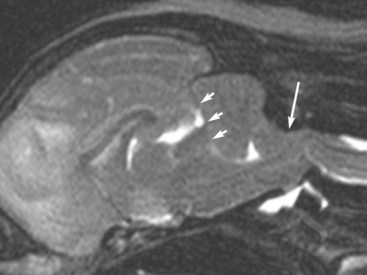
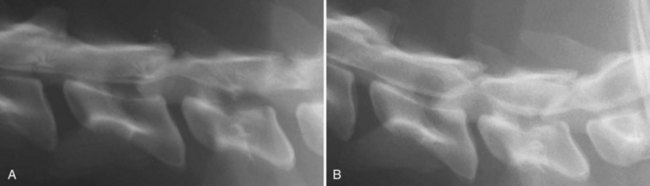
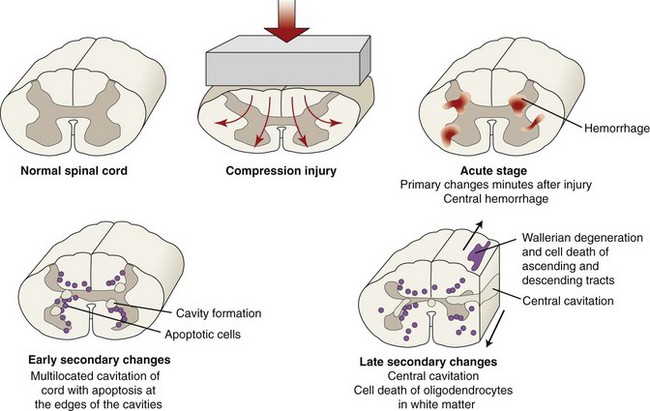
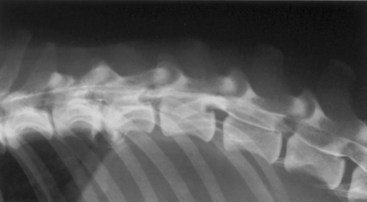
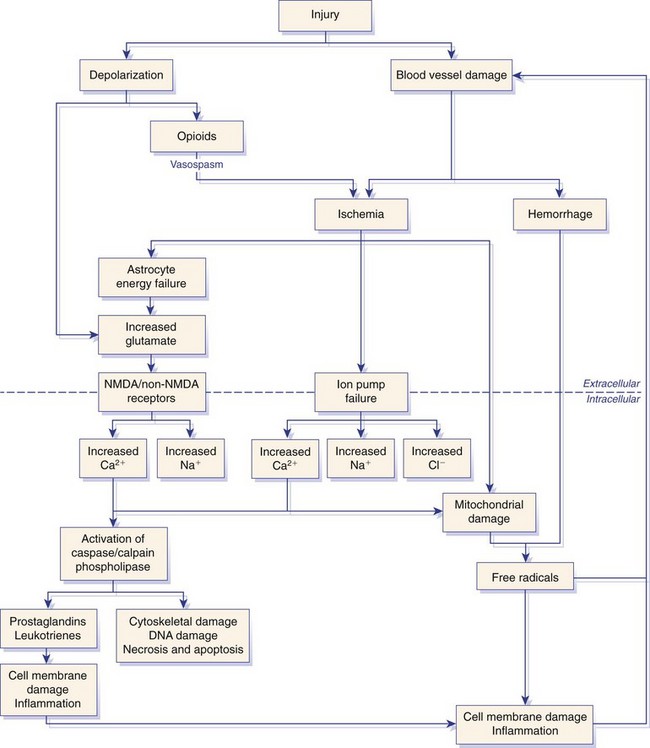
Chapter 29 In the cranium, sheets of connective tissue separate the two cerebral hemispheres (the falx cerebri) as well as the cerebellum from the cerebrum (the tentorium cerebelli). The cranium protects the brain from external trauma, and the falx and the tentorium minimize movements of the brain within the cranial cavity. Unfortunately, these protective layers can also exacerbate dysfunction caused by increased volume of the intracranial contents. Increased intracranial volume (leading to increased intracranial pressure) can be alleviated only by displacement of some of the contents. Because the front of the cranium (cribriform plate) is solid, the brain may be forced caudally either ventral to the tentorium or through the foramen magnum. Less commonly, parts of one cerebral hemisphere can be displaced laterally (i.e., ventral to the falx cerebri). Any of these movements, termed herniation, can cause brain compression, leading to well-defined neurologic syndromes and sometimes death86 (Figure 29-1). The design of the central nervous system permits transmission of electrical impulses between different points. Generation and conduction of impulses depends on the electrical excitability of nerve cell membranes.2,27,80 At rest, cell membranes maintain a negative resting potential of approximately 80 mV, with the inside of the cell negative with respect to the outside. Intracellular concentrations of potassium and extracellular concentrations of sodium are high, producing concentration gradients down which ions can move when appropriate channels open. Electrical impulses (action potentials) are generated by rapid depolarization of the membrane because of an influx of sodium ions through voltage-gated sodium channels. Action potentials arise at the axon hillock (the junction between the axon and the neuronal cell body), where excitatory and inhibitory impulses generated by afferent input to the neuron are integrated. If the sum of these impulses causes sufficient depolarization of the axon hillock, an action potential will be generated in an “all or nothing” fashion. After passage of the action potential, the membrane is repolarized by closure of sodium channels and efflux of potassium through open potassium channels. Electrolyte concentrations are returned to resting levels by active extrusion of sodium from the cell in exchange for potassium, and by potassium uptake by astrocytes. Many central nervous system axons are surrounded by myelin, a fatty envelope produced by oligodendrocytes, which allows rapid and efficient conduction over long distances while minimizing axonal diameter.2,27,38 This sheath is discontinuous, interrupted by small gaps, or nodes. The high electrical resistance and low capacitance of this myelin sheath limit transmembrane ion flux to the nodes, so that action potentials are conducted rapidly in a saltatory manner from node to node. Maintenance of a resting potential and generation and conduction of action potentials are dependent on energy (the Na+-K+/ATPase), appropriate intracellular and extracellular electrolyte concentrations, ion channel function, and myelin. Whenever the myelin sheath, oligodendrocytes, energy production, electrolyte concentrations (and therefore astrocytes), and/or ion channels are adversely affected, impulse conduction can be impaired. The central nervous system relies on a constant supply of oxygen and glucose to provide most of its high energy requirements. Efficient mechanisms have developed to match local blood flow to changing metabolic requirements (metabolic autoregulation) and to protect central nervous system perfusion from fluctuations in systemic blood pressure (pressure autoregulation) and systemic hypoxia and hypercapnia.66 Pressure Autoregulation: Perfusion of the central nervous system remains constant despite fluctuations in mean arterial pressure between 50 and 160 mm Hg60,66 (Figure 29-2) by vasodilation during hypotension and vasoconstriction during hypertension. The mechanisms by which this occurs are unclear. A myogenic mechanism is most widely accepted,141 attributing pressure autoregulation to pressure-sensitive smooth muscle cells in central nervous system blood vessels. Other theories implicate reliance on metabolite concentration.83 Any disease of the central nervous system can impair pressure autoregulation, leaving central nervous system perfusion dependent on arterial blood pressure and (in the case of the brain) intracranial pressure.60,66 Metabolic Autoregulation: At a cellular level, astrocytes play a key role in matching blood flow to neuronal activity.84 Astrocytes are able to detect changes in the chemical milieu surrounding many thousands (or even millions) of synapses, and they have contact, through their endfeet, with capillaries within the substance of the central nervous system. A very large number of signaling molecules, including nitric oxide, carbon monoxide, potassium,108 adenosine,72 glutamate, and metabolites of arachidonic acid, have been implicated in matching blood flow to metabolic requirements. As in the rest of the body, dilation and constriction of pressure vessels within the central nervous system is largely mediated via endothelial-derived factors, such as endothelium-derived relaxing factor and endothelin, although the exact identity of these factors in the central nervous system is constantly revised.29,126 In the same manner as pressure autoregulation, injury to the central nervous system impairs metabolic autoregulation. Central nervous system perfusion is extremely sensitive to changes in arterial partial pressure of carbon dioxide (PaCO2), displaying marked increases during hypercapnia and reductions during hypocapnia (see Figure 29-2). A 1 mm Hg change in arterial PaCO2 causes a 5% change in cerebral perfusion.66 Blood flow is less affected by changes in partial pressure of oxygen (PaO2) within the physiologic range, but if PaO2 falls below 50 mm Hg, central nervous system perfusion is markedly increased66 (see Figure 29-2). The potent effects of PaCO2 on blood vessel tone have important implications in brain disease. For example, injury to the brain may reduce respiratory drive, causing PaCO2 to increase, in turn inducing vasodilation of the cerebral vessels. This increases intracranial volume and therefore pressure and increases the likelihood of fatal brain herniation. Conversely, aggressive hyperventilation that reduces PaCO2 to less than 25 mm Hg causes vasoconstriction,62 potentially further compromising the injured area. Cerebral Ischemic Response and Cushing’s Reflex: Cerebral blood flow is determined by the cerebral perfusion pressure (CPP), which is defined as the mean arterial blood pressure (MABP) minus the intracranial pressure (ICP) (CPP = MABP − ICP). Factors causing a reduction in arterial blood pressure or an increase in intracranial pressure may therefore impair cerebral perfusion. Marked hypotension or elevation in intracranial pressure may reduce cerebral perfusion enough to cause ischemia of neurons in the medulla. This in turn causes a massive increase in systemic vasomotor tone to increase mean arterial blood pressure and therefore cerebral perfusion pressure. The resultant systemic vasoconstriction can be so intense that it is damaging to other organs such as the kidneys. The ensuing systemic hypertension activates baroreceptors, causing a reflex bradycardia (Cushing’s reflex).62 If cerebral perfusion pressure drops further, continued catecholamine release (as much as one thousand-fold elevations of epinephrine have been reported) can result in myocardial ischemia and ventricular arrhythmias (brain-heart syndrome).81,133 Intracranial pressure is determined by the total volume of the contents within the cranial cavity (brain, cerebrospinal fluid, and blood). Normal values for intracranial pressure in dogs and cats are in the region of 8 to 15 mm Hg.7,8 Many diseases of the brain result in increased intracranial content, thereby increasing intracranial pressure. This has serious and potentially fatal consequences because it can result in a decrease in cerebral perfusion and can ultimately cause herniation of the brain (see anatomy section).67,86 Up to a point, gradual increases in volume of intracranial components can be accommodated with minimal changes in intracranial pressure because of intracranial compliance (Figure 29-3). This consists of several mechanisms, including moving cerebrospinal fluid to the subarachnoid space of the cerebellomedullary cistern, reducing cerebrospinal fluid production, and decreasing cerebral blood flow. When these compensatory mechanisms are exhausted, intracranial pressure increases. Intracranial pressures greater than 15 to 20 mm Hg are considered abnormal, and treatment is recommended,7,8 although significant reduction in cerebral perfusion does not occur until intracranial pressure is greater than 30 mm Hg in human beings.53 A full discussion of methods of reducing intracranial pressure and maintaining cerebral perfusion pressure can be found in the Guidelines for the Management of Severe Traumatic Brain Injury.145 Surgeons should note that durotomy decreases intracranial pressure by 65%, compared with a 15% reduction from craniotomy alone.8 With the advent of magnetic resonance imaging (MRI), we now know that slow increases in intracranial pressure can result in transtentorial or transforamen magnum herniation with remarkably few associated clinical signs (Figure 29-4). However, such patients are at great risk for acute decompensation and death. The blood-brain barrier is the term used to describe the anatomic and physiologic means by which the central nervous system parenchyma is segregated from the general circulation. This microscopic barrier is formed by tight junctions between endothelial cells and by astrocyte processes, a basal lamina, pericytes, and perivascular microglia.112 Astrocytes play an important role in inducing the development of tight junctions between endothelial cells in the central nervous system.1,124 At the choroid plexuses, a blood-cerebrospinal fluid barrier is also formed, and tight junctions lie between choroid epithelial cells, rather than between capillary endothelial cells. The blood-brain barrier is selectively permeable, and free diffusion of substances through the barrier is dependent on lipid solubility, ionization, and size. Thus it restricts entry of large molecules, such as antibodies, but cytokines and certain drugs can enter the central nervous system from the circulation (Table 29-1). Specific transport systems are used for vital substances such as glucose, amino acids, and thiamine.155 Substances not only are excluded from entry to the central nervous system by the blood-brain barrier, but are actively extruded by energy-dependent efflux pumps such as p-glycoprotein.31 Mutations in the gene encoding this protein (the multi-drug-resistance gene—MDR1, now called ABCB1) are a well-known cause of susceptibility to adverse effects of drugs such as ivermectin in Collie breeds.98 The endothelial cells and perivascular microglia of the blood-brain barrier also play an important role in inflammatory responses as antigen presenting cells (see later). Table • 29-1 Antibiotic Penetration of the Blood-Brain Barrier45 Components of both innate (phagocytes and natural killer cells) and acquired (B- and T-lymphocytes) immune systems are involved in reactions within the central nervous system.13 However, the central nervous system has been termed immunologically privileged because it is relatively isolated from the immune system by the blood-brain barrier, an immunosuppressive parenchymal microenvironment, and poorly developed lymphatic drainage. The meninges and choroid plexuses do not have a blood-brain barrier and therefore can undergo inflammatory responses similar to those seen in peripheral tissues, with accumulation of neutrophils and mononuclear cells. In the normal animal, lymphocytes (which carry out immune surveillance throughout the body) have limited access to the central nervous system, and antigens derived from the central nervous system have restricted access to the general circulation. Although immune responses and inflammation have a different character within the central nervous system compared with that observed in peripheral tissues, the central nervous system is able to mount an effective immune response if activated T-lymphocytes interact with appropriate antigens within the parenchyma.102 A series of protective immunologic mechanisms expressed by the central nervous system limit entry of pathogens and other exogenous material. Expression of major histocompatibility complex molecules plus co-expression of co-stimulatory molecules (e.g., B7) is necessary for cells to act as antigen presenting cells and induce lymphocyte activation. Endothelial cells do not normally express these molecules74 and, even when induced to do so, are not able to induce T-cell proliferation.14 Cell adhesion molecules, which are required for lymphocytes to adhere to and then cross blood vessel walls, are normally expressed only at low levels on endothelial cells (thus forming part of the barrier to lymphocyte entry into the central nervous system) but are rapidly upregulated in response to appropriate cytokines.91 The next cells encountered by activated T-cells surveying the central nervous system are the perivascular macrophages, which constitutively express major histocompatibility complex class II molecules and are competent antigen presenting cells.19 Microglial cells, which are the resident immune and phagocytic cells of the central nervous system,49,50,148 may also express major histocompatibility complex molecules, although this varies between species.49,131 However, they are able to rapidly upregulate their expression of major histocompatibility complex class II molecules and furthermore co-express B7, permitting their participation in immune responses within the parenchyma.13,14 Whether astrocytes function to promote or suppress immune responses has not been fully answered, and it is likely that their responses vary in different phases or types of pathologic response.14 Stem Cell Populations in the Adult Nervous System: In the past two decades, discovery of stem cell populations within the adult mammalian nervous system has reversed the previous belief that neurogenesis did not occur in adult mammals.48,78,122 A neural stem cell is defined as a cell that is self-renewing (a theoretically unlimited characteristic), proliferative, and multipotent for neurectodermal lineages (glial and neuronal).139 Cells that are precursors to neurons, astrocytes, or oligodendrocytes (i.e., lineage specific) are known as precursor or progenitor cells. Constitutive neurogenesis occurs in the adult nervous system in two locations within the brain: the subventricular zone/olfactory system and the dentate gyrus of the hippocampus. Neogenesis has been proposed in numerous other areas (e.g., neocortex, spinal cord) by some, but not all, workers in the field. Stem cell populations have also been isolated and grown in vitro from many other niches within the central nervous system. Such findings have raised the possibility of using these endogenous stem cell populations to regenerate lost or damaged neural tissue. Relatively little is currently known about neural stem cells in dogs and cats. Recently, the subventricular zone/olfactory system was demonstrated to contain stem cells in adult dogs and cats,92 and neural stem cells were isolated from several regions of adult dog brains.93 Many different diseases can affect the central nervous system, but for simplicity, their effects have been subdivided into a few basic categories: contusive, compressive, inflammatory, vascular, metabolic, toxic, degenerative (Table 29-2), and neoplastic. Although brain trauma is a major cause of morbidity and mortality in man, relatively little has been written on the subject in the veterinary literature.35,36 Spinal cord trauma is more common (see Table 29-2), particularly because vertebral column mobility can result in repeated spinal cord contusion (Figure 29-5). This may explain the clinical improvement that can be produced by fusion of two vertebrae in equine cervical stenotic myelopathy, without concomitant decompression of the spinal cord.104 Contusion of the brain or spinal cord causes primary mechanical damage to the central nervous system, initiates a chain of biochemical events that cause neuronal and glial cell necrosis and apoptosis (secondary injury), and incites an inflammatory response. Secondary consequences of the initial injury continue to develop over a prolonged period (Figure 29-6). Brain: Trauma to the brain typically results in a combination of focal and diffuse pathologic changes. Focal brain injuries alone are unusual and result from static loading (defined as forces applied to the head during a period longer than 200 ms). More commonly, the head suffers a dynamic injury as the result of an impact (e.g., a fall) or of rapid acceleration and/or deceleration. These forces cause movement of the brain within the cranial cavity and hence both focal and diffuse pathologic changes.44 Diffuse brain injury refers to widespread damage to gray and/or white matter caused by brain swelling, hypoxia, and, most important, diffuse axonal injury.56,119 Diffuse axonal injury results from inertial forces causing membrane damage to axons throughout the brain, allowing unregulated sodium entry and depolarization, which initiates the secondary effects described later. Diffuse axonal injury is the cause of posttraumatic coma and death in which a mass lesion is not identified.56,134 Spinal Cord: Primary mechanical damage varies widely in severity, ranging from complete transection of the spinal cord in displaced vertebral fractures to minimal injury in disc herniations. This variability is paralleled by the observed difference in prognosis for the return of pelvic limb function in dogs that present paraplegic with no deep pain sensation resulting from displaced vertebral fractures26 (Figure 29-7) compared with that in dogs that have sustained a similar degree of neurologic dysfunction following intervertebral disc herniation.130 The concept of secondary injury following trauma to the central nervous system is now well established and reviewed.36,56,109,143 Traumatic injuries (and ischemic injuries, see later) initiate a series of biochemical and metabolic changes that cause neuronal and glial cell death (Figure 29-8). This process starts within seconds of injury and, although most cell necrosis occurs within the first 24 hours of injury, apoptotic cell death may continue for weeks.32 Traumatic injury initiates secondary events by direct injury to neuronal, axonal, and glial cell membranes and blood vessels (Figure 29-9, A). A recent study51 has revealed the important role played by de novo gene expression in expanding the region of hemorrhage after experimental spinal cord injury. The transient membrane potential family of genes encode cation channels, and in the case of transient membrane potential 4 and 5, monovalent cation channels. Within 3 hours of injury, transient membrane potential 4 is widely upregulated, especially in endothelial cells, and blockade of this effect (e.g., in knockout mice) is associated with reduced zones of hemorrhage, reduced lesion volume, and improved outcome. Transient membrane potential 4 therefore appears to play a central role in ongoing hemorrhage after injury. The consequent decreased perfusion of the injured area reduces the energy supply to neurons and glial cells, causing ion pumps to fail or reverse; subsequent cell membrane damage causes increased permeability. As a result, intracellular levels of sodium, chloride, and calcium ions increase, upsetting osmotic balance and causing cellular swelling (cytotoxic edema) (see Figure 29-9, B). The increase in intracellular sodium and calcium is perpetuated by an increase in extracellular glutamate (and, to a lesser extent, aspartate) concentration, which, in excessive concentrations, can cause both neuron and oligodendrocyte death (excitotoxicity).96,164 This occurs because extracellular concentrations of glutamate are normally regulated by efficient active uptake by astrocytes.125 Mechanical damage to neurons, plus local energy failure, results in increased neuronal release and decreased astrocytic uptake of glutamate, elevating the extracellular concentrations to toxic levels. Interaction of glutamate with N-methyl-D-aspartate (NMDA), alpha-amino-3-hydrocy-5-methyl-4-isoxazole propionic acid (AMPA), and kainate receptors causes a rapid increase in intracellular sodium and a more gradual increase in intracellular calcium. Increased intracellular calcium concentration does the following: Figure 29-9 A, Transverse section of thoracolumbar spinal cord from a Cocker Spaniel that had suffered an acute intervertebral disc herniation 24 hours previously. The dog presented as paraplegic with no deep pain sensation. Hemorrhages can be seen within the spinal cord. The basic structure of the spinal cord is still intact (compare with Figure 29-12). (Hematoxylin ×20.) B, Transverse section of the spinal cord gray matter 30 minutes after thrombosis of the spinal cord vasculature (arrow points to a thrombosed blood vessel). The neuropil is vacuolated as a result of intracellular swelling (cytotoxic edema). (Toluidine blue ×350.) (From Olby N, Jeffery N: Pathogenesis of diseases of the central nervous system. In Slatter DH, editor: Textbook of small animal surgery, ed 3, Philadelphia, 2003, Saunders/Elsevier.) 1. Activates intracellular proteases such as calpains and caspase, which destroy the cytoskeleton and chromosomes and initiate programmed cell death154 2. Activates phospholipase A2, thereby producing eicosanoids and initiating an inflammatory response11 3. Binds intracellular phosphates, further depleting the cell of energy sources161
Pathogenesis and Physiology of Central Nervous System Disease and Injury
Background
Physiology
Regulation of Central Nervous System Perfusion
Intracranial Pressure
The Blood-Brain Barrier
GOOD PENETRATION
MODERATE PENETRATION WITH MENINGITIS
POOR PENETRATION
Third-generation cephalosporins
Fluoroquinolones
Tetracyclines: especially doxycycline and minocycline
First- and second-generation cephalosporins
Metronidazole
Erythromycin
Aminoglycosides
Sulfonamides
Penicillins
Clindamycin
Chloramphenicol
Rifampin
Vancomycin
Trimethoprim
Physiology of the Immune System of the Central Nervous System
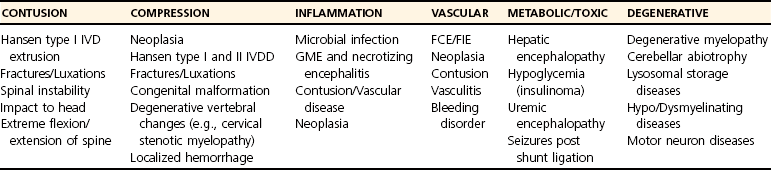
Lesion Pathogenesis
Contusion
Primary Mechanical Damage
Secondary Injury Mechanisms
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Pathogenesis and Physiology of Central Nervous System Disease and Injury
Only gold members can continue reading. Log In or Register to continue
