(1)
Department of Immunology, Genetics, and Pathology, Uppsala University, Uppsala, Sweden
Abstract
Oligodendroglial tumors are primary tumors of the central nervous system that largely affect adults. The cell of origin is undefined, but the tumors display many features reminiscent of oligodendrocytes or oligodendrocyte progenitor cells. Here, we briefly recapitulate the history of oligodendroglial tumor research, discuss the current knowledge concerning the biology of oligodendroglial tumors, and thoroughly review the various mouse models that have been used and that are currently in use to study oligodendroglial tumor development.
Key words
OligodendrogliomaOligodendrocyteOligodendrocyte progenitor cell (OPC)Platelet-derived growth factor receptor alpha (PDGFRA)Epidermal growth factor receptor (EGFR)CNPaseOLIG2NG2Mouse model1 Introduction
Oligodendrogliomas display features of oligodendrocytes in their histological appearance. Oligodendrocytes are cells of the central nervous system (CNS) with main purpose to myelinate neurons. In this chapter, experimental models of oligodendroglioma are in focus. Histopathology and molecular genetics of human oligodendroglioma are described, and brief accounts of early oligodendroglioma models and more detailed descriptions of current models presented. Oligodendroglial tumors include according to the current WHO classification (Fig. 1a–d): oligodendroglioma (WHO grade II), anaplastic oligodendroglioma (WHO grade III), oligoastrocytoma (WHO grade II), and anaplastic oligoastrocytoma (WHO grade III). For simplicity, we refer collectively to all types of oligodendroglial tumors as oligodendroglioma. In those instances where we specifically mean the clinical subtype oligodendroglioma we add grade II where applicable.
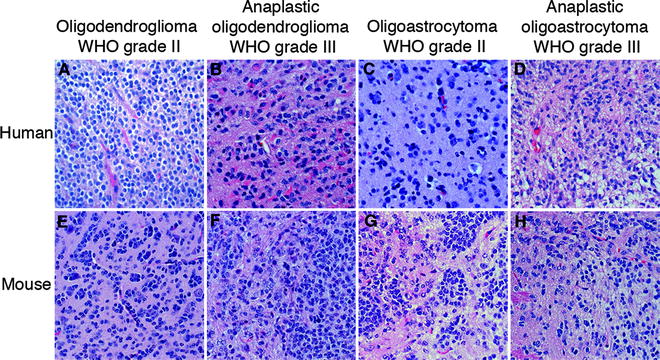
Fig. 1.
H&E stained sections of the various types (oligodendroglioma, anaplastic oligodendroglioma, oligoastrocytoma, anaplastic oligoastrocytoma) and grades (II and III) of (a–d) human oligodendroglial tumors and (e, f) PDGF-B-induced mouse oligodendroglial tumors from the Ctv-a model (142).
2 Classification and Biology of Oligodendroglioma
2.1 Historical Notes
In the early twentieth century, neurosurgeons and pathologists began describing glial tumors with oligodendroglial histology establishing them as a separate tumor subtype. Neuroglia (“nerve glue,” the cells that glue together the nerves) were at this time described as supportive cells of the CNS with elaborate fibrillae which today are known as astrocytes. Also neuroglial cells lacking fibrillae had been observed and were first described by W. F. Robertsson in 1899 as a different subset of neuroglia (1). These were portrayed as small process-bearing cells present throughout the gray and white matter that could be selectively stained using a platinum impregnation technique. They were believed to be derived from the mesoderm and were called mesoglia when they in fact were the first oligodendrocytes to be described. In 1921, del Río-Hortega identified oligodendroglia to be of ectodermal origin and renamed these cells oligodendrocytes. In 1924, Bailey and Cushing discussed how cell-specific chemical color reactions, such as the platinum impregnation technique used by Robertsson et al., could be used to identify the corresponding normal cell type that would most resemble that of a particular cancer cell (2). The current belief was that the supporting tissue of the CNS constituted neuroglia (astrocytes), microglia, and oligodendroglia (also called oligocytes), and that tumors of the CNS were composed of and putatively originated from these supporting cells rather than the nervous tissue itself. Tumors with oligodendroglial-like cells were either called oligocytomas or oligodendrogliomas. In 1929, Bailey and Bucy described a set of 13 oligodendrogliomas comparing the tumor cells to oligodendroglia (3).
The comparisons of morphological characteristics of oligocytes and astrocytes in normal tissue and cerebral tumors were the subject for intense investigations by Eugenia R.A. Cooper in the 1930s (4). Cooper described an oligocytoma as containing closely packed round “empty” cells, a tissue processing artifact later referred to as perinuclear halo. Other characteristics such as proliferation and migration of endothelial cells and presence of enlarged astrocytes (later termed “reactive astrocytes”) infiltrating the tumor area were described by Bailey and Cooper and others. When reading Cooper’s report it is evident that he did not believe that an oligocyte could change into an astrocyte or develop into an astrocytoma and vice versa. Rather, he thought that oligocytomas and astrocytomas strictly arose from their normal cellular counterparts. The possible cell of origin for gliomas has been heavily debated during the years and today we know that transitions between different cellular lineages can in fact occur, at least in experimental glioma (5–9).
The relationship between precursor cells of oligocytes and astrocytes and the cellular origin of oligodendroglioma was also discussed by Penfield who stated that the characteristics of an oligocytoma cell was that of an oligodendroblast, a less differentiated cell (10). He was supported by del Río-Hortega, reviewed in (11), who described the tumor cells of oligodendroglioma as incompletely differentiated, since myelinated fibers were absent. Many of these early observations and theories would later prove to be correct.
2.2 Histological Characteristics
Oligodendrogliomas comprises cells with oligodendrocyte appearance having small to slightly enlarged nuclei, with compact chromatin surrounded by a small amount of eosinophilic cytoplasm. As mentioned above, due to a tissue processing artifact oligodendroglioma cells typically display a clear cytoplasm around a central spherical nucleus making them look like “fried eggs” (Fig. 1a). This occurs when the tissue is formalin-fixed which causes degeneration due to acute swelling leaving an empty space between the nucleus and the cellular membrane, a feature called perinuclear halo, giving the tissue a honeycomb appearance. Tumor cells are round in shape and relatively homogenous within the tumor. As for all gliomas, they infiltrate the normal brain parenchyma irrespective of malignancy grade. A hallmark of oligodendrogliomas is the endothelial cell proliferation and migration seen as “chicken-wire”-like small, branching capillaries (Fig. 1a). Neoplastic oligodendrocytes commonly cluster around neurons, “perineuronal satellitosis,” or around vessels forming “pseudorosettes.” They preferentially migrate along white matter tracts or vessels spreading diffusely within the brain and across the hemispheres.
2.3 Grading, Markers, and Prognosis
Oligodendrogliomas are commonly classified using guidelines from the World Health Organization (WHO), most recently updated in 2007 (12). Currently, oligodendroglial tumors are classified into grade II (oligodendroglioma, oligoastrocytoma) and grade III tumors (anaplastic oligodendroglioma, anaplastic oligoastrocytoma) (Fig. 1a–d). Grade II tumors are composed of homogenous, diffusely infiltrating, round cells with moderate cell density. Nuclear atypia and occasional mitotic figures may be present, but significant mitotic activity, considerable vascular proliferation, and necrosis are signs of tumor progression into a grade III tumor. Grade III tumors are classified by significant hypercellularity and pleomorphism. Unlike astrocytic tumors, there is no grade IV classification for oligodendroglial tumors. Tumors with characteristics of grade IV glioma [glioblastoma (GB)] arising from a previously diagnosed oligodendroglial tumor are currently graded as “glioblastoma with an oligodendroglial component.” However, it is not clear if these tumors represent a subset of GBs with differential origin, prognosis, and treatment requirements.
There are no cellular markers that are exclusive for oligodendrogliomas. Many oligodendroglial markers are expressed by astrocytic tumors and vice versa. Some markers expressed by normal oligodendrocytes are however more predominantly expressed in oligodendroglial tumors such as OLIG2 (13–16), chondroitin sulfate proteoglycan NG2 (17), SOX10 (18–20), and NKX2-2 (20, 21). Also, 1p/19q loss is rare in astrocytomas and is used in the clinic to aid in the diagnosis of oligodendrogliomas. Loss of 1p or 1p/19q is associated with better prognosis for patients with grade II and possibly also grade III oligodendrogliomas (22–29). This is likely due to an increased sensitivity to chemotherapy of these tumors (26–28), a notion further supported by several gene expression profiling analyses that have identified oligodendrogliomas carrying 1p/19q codeletions to be associated with a favorable prognosis (30, 31).
Oligodendroglioma and oligoastrocytoma respond well to combined treatment using procarbazine, lomustine (CCNU), and vincristine, in short PCV treatment (32–35). They are also sensitive to the alkylating agent temozolomide (36). The prognosis for patients diagnosed with oligodendroglioma varies from 1 to 10 years with LOH 1p/19q being a significant positive prognostic factor (37, 38).
2.4 Core Signaling Pathways
The spectrum of mutations in oligodendroglial tumors overlap to large extent with those found in astrocytic tumors but there are also some distinct differences. Figure 2 shows a summary of the most frequently mutated genes in oligodendroglioma that are discussed below.
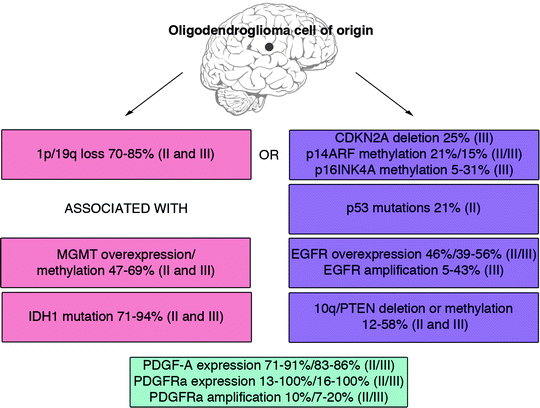
Fig. 2.
The frequency of core mutations in human oligodendroglial tumors of grade II and III. There appear to exist at least two different paths to develop oligodendroglioma. One displays loss of 1p/19q (37, 39–43), MGMT overexpression/promoter methylation (49, 50) and IDH1 mutation (55–57). The other path exhibits CDKN2A deletion/methylation (50, 61), p53 mutation (49, 64), EGFR amplification (25, 68, 70), and loss of 10q (25, 68). PDGF-A/PDGFRa overexpression or PDGFRa amplification seem to occur independent of 1p/19q status (82–84).
Combined loss of chromosomes 1p and 19q is the most common genetic alteration and is considered to be a hallmark of oligodendroglioma. While rare in astrocytic tumors single or codeletion of chromosomes 1p and 19q can be found in 70–85% of oligodendrogliomas (37, 39–43). As mentioned above, the loss of 1p/19q can be used both as a diagnostic and prognostic tool (37, 38). Since 1p/19q deletions can be found in both grade II and grade III tumors, they are believed to be early events.
DNA repair protein O6-methylguanine-DNA methyltransferase (MGMT) is often overexpressed in oligodendrogliomas causing treatment resistance to alkylating agents such as temozolomide, procarbazine, and CCNU (44, 45). Hypermethylation of MGMT leads to lower expression of the protein and thereby a better response to chemotherapy and a favorable prognosis for patients with oligodendroglioma grade II (46–50) and anaplastic oligodendroglioma grade III (51). In vitro, MGMT expression levels are lower in oligodendrocytes than in astrocytes which leads to higher chemosensitivity of oligodendrocytes to BCNU (52), suggesting that oligodendrocytes express low levels of MGMT and that overexpression is associated with tumorigenesis. Interestingly, hypermethylation of MGMT is associated with the loss of 19q and 1p/19q (50, 53, 54).
Heterozygous mutations of isocitrate dehydrogenase 1 (IDH1) at amino acid 132 (R132) result in the loss of the enzyme’s ability to catalyze the conversion of isocitrate to α-ketoglutarate. Mutations at this residue is an early event in oligodendroglioma (55), since it is frequently found in both grade II and III oligodendroglial tumors (56, 57). In oligodendroglioma grade II, there is a correlation between IDH1 mutations and 1p/19q loss (55). This link was supported by another study, which showed that presence of IDH1 mutations correlated with both 1p/19q loss and MGMT promoter methylation, and could be used as a prognostic factor for overall survival (58).
The CDKN2a locus encodes tumor suppressor proteins p16INK4a and p14ARF, both of which are frequently methylated and occasionally deleted in oligodendrogliomas (25, 50, 59–61). Alterations of p14ARF are more common in anaplastic oligodendroglioma than in oligodendroglioma grade II (62), but no correlations to survival have been reported. Decreased expression of p16INK4a predicts poor survival (63), and loss of 1p/19q and CDKN2A alterations appear to be mutually exclusive (37).
While common in astrocytic tumors mutations of TP53 are infrequent in oligodendroglial tumors (49, 64). Similar as for CDKN2A alterations, oligodendroglial tumors rarely have concomitant TP53 mutation and 1p/19q loss (42, 65).
Epidermal growth factor receptor (EGFR) overexpression, mutation, and amplification occurs less frequently in oligodendroglial than in astrocytic tumors (66, 67), and these alterations can be associated with poor prognosis (25, 68, 69). Many grade III tumors exhibit EGFR overexpression that can be coupled to EGFR amplification (66, 70), but there are also oligodendroglial tumors of both grade II and III that lack EGFR amplification but still exhibit overexpression of EGFRs (71). EGFR amplification has been found to be mutual exclusive to 1p/19q deletions (25, 68, 70).
Alterations affecting PTEN are very common in astrocytic tumors and occur in 74–87% of glioblastomas (72–74) and in 37–75% of anaplastic astrocytomas (73–75). PTEN alterations are also common in oligodendrogliomas where they occur in up to 48% of the tumors (37, 42, 76, 77). Such alterations involve mutations, promoter methylations, and 10q deletions causing loss of PTEN expression/function (25, 46, 77–81). Loss of 10q seems to be mutual exclusive to 1p/19q deletions (25, 68).
Platelet-derived growth factor-A (PDGFA) and PDGF receptor-A (PDGFRA) are commonly expressed in oligodendroglial tumors and appear to be an early event (82, 83). A subset of tumors also shows amplifications in the PDFGRA gene (83, 84).
High-grade gliomas have been grouped into three subclasses based on their molecular expression signatures; proneural, mesenchymal and proliferative (31), where all oligodendroglioma grade III tumors were stratified to the prognostically favorable proneural subclass. In a different investigation, gliomas of all grades and histologies were classified based on activation of PDGFRA, EGFR, and NF1 (85). In this study, two oligodendrogliomas grade II and two oligoastrocytomas were included, and the result indicated that oligodendroglial tumors could be overrepresented in the group of tumors driven by PDGF signaling. The Cancer Genome Atlas Research Network (TCGA) presented a gene expression signature classification of glioblastomas (grade IV astrocytomas) that described four different subtypes of glioblastoma (neural, proneural, classical, and mesenchymal) (86) that displayed reasonable similarities with the molecular subtypes described by Phillips et al. This subclass classification was used in a very recent study that included 43 oligodendrogliomas which showed that the majority of oligodendroglial tumors belonged to the proneural group as defined by TCGA. These were also associated with a significantly better prognosis than oligodendrogliomas stratified to the classical or mesenchymal subgroups (87). Loss of 1p/19q was common in the oligodendrogliomas and was overrepresented in those tumors belonging to the proneural group. Taken together, we can conclude that oligodendrogliomas appear to be driven by somewhat different signaling pathways than astrocytomas, and that there seem to be at least two independent routes leading to oligodendroglioma development.
3 Cellular Origin of Oligodendroglioma
As mentioned above, oligodendrogliomas have been named after the cell type that they share most histological and morphological resemblance to. Notably, the cell of origin for oligodendroglioma is still unknown. As described previously, already in the 1920s and 1930s neuropathologists and neurosurgeons debated whether the histology of glial tumor cells reflected their cellular origin, the relationship between oligodendrocytes and astrocytes, and the existence of a common or shared progenitor cell (4, 10, 11). During the late twentieth century, many studies were focused on characterizing markers expressed by human oligodendrogliomas, and the result showed that they indeed shared many antigens with normal oligodendrocytes and oligodendrocyte progenitor cells (OPCs). Some markers of OPCs that are commonly expressed in oligodendrogliomas are NG2 (17, 88), PDGFRA (17, 88), OLIG2 (13, 14, 16, 89), GalC in mouse and human, respectively (90, 91), MBP (92), A2B5 (91, 93), and Leu-7 in rat oligodendrogliomas (94). Antigens of mature oligodendrocytes, for example proteins associated with myelination such as proteolipid protein (PLP) and 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNPase), are only rarely expressed in oligodendrogliomas (95), confirming the observation by del Río-Hortega (11) that oligodendrogliomas exhibit OPC characteristics. Thus, the OPC with its high capacity for self-renewal seem to be in a favorable state for promoting neoplastic transformation, which does not necessarily mean that OPCs are the (sole) cell of origin for oligodendroglioma.
It should be noted that both astrocytic and/or neuronal markers are at times expressed in oligodendrogliomas (96, 97). This could point to that oligodendrogliomas and astrocytomas in some instances may have a shared progenitor cell or merely illustrate the disturbed differentiation potential of glioma cells. Genetic analyses suggest that oligodendrogliomas and oligoastrocytomas have a common cellular origin due to their similar genetic profiles such as loss of 1p and 19q (98), and that the astrocytic component represents the skewed differentiation potential of the tumor cells.
There are studies supporting that GBs with a significant oligodendroglial contribution might have developed from an oligodendroglioma grade II or III rather than from an astrocytoma. GBs without TP53 mutations or EGFR gene amplification could be oligodendroglial GBs, since oligodendrogliomas rarely carry these genetic alterations as suggested by (99). Consequently, the histopathology of oligodendroglioma does not formally prove the cellular origin of these tumors.
4 Animal Models of Oligodendroglioma
Gliomas cannot be diagnosed until a tumor is present, and most gliomas are diagnosed at a late stage of the disease, which makes it difficult to study the mechanisms of tumor initiation in human biopsy material. Furthermore, due to ethical considerations clinical trials of novel drugs have to be preceded by preclinical trials in experimental models. Animal models of oligodendroglioma can help improve our knowledge of genetic, molecular, cellular and systemic mechanisms that contribute to tumor development, and be used in preclinical trials to identify promising candidate drugs.
4.1 Properties of a High Quality Oligodendroglioma Model
In our view, there are three basic features that need to be fulfilled in order to be regarded as a relevant and adequate animal model for oligodendroglial tumors: (1) The histopathology of the experimental tumor should recapitulate the histological characteristics of the corresponding type and grade of human oligodendroglioma. (2) The genetic alteration(s) should mimic signaling pathways activated in human oligodendroglial tumors. (3) The tumor should be orthotopic so that it develops in the proper microenvironment. Also, there are several additional properties of an experimental model that are highly desirable: (4) A defined cell of origin. (5) Temporal control of tumor initiation. (6) High tumor incidence and penetrance and short tumor latency. (7) Possibility to model different grades of oligodendroglial tumors.
The experimental models presented here have all generated tumors that have been classified as oligodendroglioma-like by their authors at the time of publication. Many of these models have produced tumors of various histopathologies, i.e., oligodendroglioma, oligoastrocytoma, and astrocytoma. This could be due to that astrocytic and oligodendroglial tumors share many genetic alterations but could also reflect that these different tumor types may in some instances arise from the same cell type. Also of note is that there have been continuous revisions of the WHO guidelines for classification of gliomas; hence, some tumors that were earlier classified as oligodendroglial tumors might not meet the criteria of today.
4.2 Early Oligodendroglioma Models
Early studies in the beginning of the twentieth century were focused on describing spontaneous oligodendroglial tumors in various animal species such as dog (100–102), cat (100, 103), rat (104–107), horse (100), and cow (100, 108). The first experimentally induced oligodendrogliomas were published in the 1960s where the Rous Sarcoma Virus (RSV) was used as the initiating agent.
4.2.1 RSV-Induced Oligodendrogliomas
RSV was the first oncovirus to be discovered by Peyton Rous in 1911, which awarded him the Nobel Prize in 1966. RSV contains the oncogene v-src (viral-sarcoma) that is derived from the protooncogene c-Src encoding an intracellular tyrosine kinase protein (109). v-Src lacks the C-terminal inhibitory phosphorylation of Tyr-527, present in c-Src, leading to constitutive activation and transformation of cells (110). In the 1960s and 1970s RSV was extensively used to induce brain tumors in dogs (111–113), mice (114), rabbits (115), and other mammals. Some of these tumors resembled oligodendroglioma while most were histologically similar to other types of glioma. This could be due to that RSV could not infect the appropriate target cells for oligodendroglioma and/or that v-Src signaling did not activate the pathways required for oligodendroglioma development. The low frequency of oligodendroglioma made this a poor model.
4.2.2 Chemically Induced Oligodendrogliomas
Various chemical agents have been used to induce experimental gliomas. The first and most commonly used chemical agent to induce gliomas was nitrosourea, a DNA alkylating agent that has cytotoxic, mutagenic, and carcinogenic effects (62). Nitrosourea can pass the blood–brain barrier, and glioma-like brain tumors were induced in both rats (116) (117, 118) and rabbits (119) by intravenous, intraperitoneal, or oral administration, e.g., the widely used C6 rat glioma cell line was established from a glioma induced by ethylnitrosurea (116). The CNS tumors induced resembled histologically human oligodendrogliomas, astrocytomas, neurinomas, and ependymomas, sometimes with intermixed cell types. For many years these models were used to study the histological and morphological composition of glial tumors (120–122). Oligodendroglial-like tumors (in minority to astrocytic tumors) were also induced in rats by the combination of dietary lead subacetate and 2-acetylaminofluorene (123), and by embryonal distribution of methanesulfonate (124). None of the chemically induced models generated a high frequency of oligodendrogliomas; the majority of induced tumors were in fact astrocytoma-like. Other major disadvantages were the random and unspecific genetic alterations caused by the agents and the undefined target cells making tumor induction unpredictable and experiments difficult to reproduce.
4.2.3 Transplanted Oligodendrogliomas
In the 1950s, it became feasible to maintain cells in culture as the knowledge of their nutritional and environmental requirements expanded (125). The first transplantation models using cultured glioma cells were published in the 1960s (126). However, most cell lines have been established from high-grade astrocytomas, such as the C6 rat glioblastoma cell line (116), and there are very few mouse oligodendroglioma cell lines (G26-24 (127)) and human oligodendroglioma cell lines (e.g., IN/157 (128), HOG7 (129), TC620 (130), MO3.13 (131), KG-1C (132)) described. Recently, two anaplastic oligodendrogliomas with complete loss of 1p/19q have been explanted and maintained as neurospheres in culture (133). Cultured cells have shown to have characteristics of CSCs, and one of the cell lines (BT088) could initiate tumors similar to anaplastic oligodendroglioma in immunocompromised mice.
One major advantage with transplanted glioma models is that they are highly reproducible with regard to tumor incidence, latency, growth rate and location given that an established, transplantable cell line is being used. A weakness of grafted tumors has been that they often do not recapitulate the invasive growth of human oligodendroglioma. This has lately been greatly improved by the use of glioma-derived cancer stem cells for transplantation. When grafted these cells generate tumors with histopathology highly similar to the primary tumor, which make these models much more relevant for tumor biology studies and preclinical testing. However, so far very few CSC lines have been established from oligodendrogliomas (133). An additional limitation of transplanted models is that the role of cellular origin and underlying mechanisms of tumor initiation and progression cannot be studied.
5 Genetically Induced Oligodendroglioma Models
These are the experimental oligodendroglioma models most commonly used today. Table 1 presents a summary of the contemporary models generating oligodendroglioma-like tumors.
Table 1
A list of the most common contemporary experimental models of oligodendroglioma development
Model system | Animal species | Target cell | Oncogene | Tumor suppressor gene | Histopathology | References |
|---|---|---|---|---|---|---|
Transplanted CSCs derived from OG | Mouse | NA | NA | 1p19q deletion | OIII | Kelly (2010) |
RCAS/TV-A | Mouse | GFAP | PyV-mt | wt | OII-III, AIII | Holland (2000) |
RCAS/TV-A | Mouse | GFAP or Nestin | PDGF-B | wt, Cdkn2a−/− | OII-III, AII-IV | Dai (2001) |
RCAS/TV-A | Mouse | GFAP or Nestin | PDGF-B | wt, Ink4a−/−, Arf−/−, Cdkn2a−/− | OII-III | Tchougounova (2007) |
RCAS/TV-A | Mouse | CNPase | PDGF-B | wt | OII-III, OAIII | Lindberg (2009) |
MoMuLV | Mouse | Unknown | PDGF-B | wt | OII-III, AII-IV | Uhrbom (1998) |
MoMuLV | Mouse | Unknown | PDGF-B | p53−/−, Cdkn2a−/− | OII-III, AII-IV | Hesselager (2003) |
transgene | Mouse | GFAP | V12Ha-Ras + EGFRvIII | wt | OII, OAII | Ding (2003) |
transgene | Mouse | S100β | v-erbB | wt, Cdkn2a+/−, p53+/− | OII-III | Weiss (2003) |
transgene | Rat | S100β | v-erbB | wt | OII-III, AIV | Ohgaki (2006) |
5.1 Retroviral Models of Oligodendroglioma
Retroviruses (e.g., Avian leukosis viruses such as RSV, Murine leukemia viruses, Lentiviruses) confer stable transduction of genes into the host genome and have been widely used to study the significance of specific genetic alterations in glioma and many other tumor types. Common for all retroviral models is that the target cell needs to be dividing for retroviral infection to occur.
Eric Holland and Harold Varmus established the first RCAS/TV-A tumor model system (134, 135) that has subsequently been used to model many other tumor types outside the CNS (136–139). The RCAS/TV-A model allowed for controlled and specific transduction of exogenous genes into distinctive glial cell populations of the brain. RCAS is a plasmid originally derived from RSV that was modified to harbor an exogenous gene (140). RCAS viruses are propagated in DF-1 chicken cells and can only infect cells expressing the avian tv-a receptor, required for infection of subgroup A Avian leukosis viruses, that is not present in the mammalian genome. Two transgenic mouse lines expressing tv-a in a cell type or tissue specific manner were generated using the nestin or GFAP (Glial fibrillary acidic protein) promoters, respectively (134, 135). The Nestin promoter will direct infection by RCAS to neural/glial stem cells in the Ntv-a line, and the GFAP promoter to astrocytes and neural stem cells of the subventricular zone in the Gtv-a line (141). Recently, our lab has published a third tv-a transgenic mouse line for the study of CNS tumors, where the 2′, 3′-cyclic-nucleotide 3′-phosphodiesterase (CNPase) promoter was used to control tv-a expression (142) and target it to OPCs. Directed infection can take place by intracerebral injection of RCAS viruses in neonatal or adult mice and the tumor will develop orthotopically in a normal microenvironment. Some advantages with the RCAS/TV-A model over the early models described are that the cellular origin, genetic alterations and the time of induction can be controlled. The RCAS virus is replication-incompetent in mammalian cells, which will limit the contribution of insertional mutagenesis. The different tv-a transgenic mouse lines have been extensively used to model many types and grades of glioma. By infecting the three tv-a lines with RCAS-PDGF-B, oligodendrogliomas grade II can be reproducibly induced (Fig. 1e) (6, 142, 143). Occasional oligoastrocytomas also developed in the Ctv-a mice (Fig. 1g) (142). Through cross-breeding the tv-a transgenic mouse lines with mice carrying targeted deletions of the Cdkn2a locus, the separate roles of tumor suppressors p16Ink4a and p19Arf in PDGF-B could be studied. Loss of p19Arf have been found to be the major suppressor causing enhanced incidence and malignancy, producing a high frequency of grade III oligodendroglioma (Fig. 1f, h) and glioblastoma in all three mouse lines (143, 144). Thus, by using tv-a transgenic mice cross-bred with Arf−/− mice high-grade oligodendrogliomas can be robustly generated. The loss of other cell cycle inhibitors such as p21Cip1 (145) and p27Kip1 (146, 147) have also been studied in PDGF-B-driven oligodendroglioma development, as well as the role of many putative glioma-causing genes/pathways such as SOX10 (19), IGFBP2 (148, 149), SOX5 (150), nitric oxide-induced signaling (151), and DNA damage response genes (152). The PDGF-induced Ntv-a model has also been used in preclinical investigations, e.g., to analyze the effects of the AKT inhibitor perifosine (153), and the antiangiogenic/immune-modulating histidine-rich glycoprotein (HRG) (154).
Moloney Murine Leukemia Virus (MMLV)-based vectors have also been widely used to model many different types of tumors, including oligodendroglioma, in mice. MMLV will deliver the exogenous gene in an undefined manner infecting most murine cell types. The first oligodendroglioma-like tumor generated by MMLV infection came from infection of fetal rat brain cells by a replication-incompetent MMLV encoding ras and myc oncogenes that were subsequently transplanted into the brains of syngeneic hosts (155). A few years later, our lab used a combination of replication-competent and replication-incompetent MMLV carrying the human PDGF-B cDNA that were injected into the brains of neonatal mice (156, 157). The mice developed tumors with a high incidence and many showed a histopathology similar to human oligodendroglioma. The model was made with the purpose to be used as a tool to identify retrovirally tagged cancer-causing genes complementing PDGF-B in glioma development and several novel and known brain tumor loci were subsequently identified (19, 150, 158–161). MMLV vectors have been used to transduce mouse neural stem cells and astrocytes in vitro with a wild type or constitutively active EGFR mutant, EGFRvIII (in-frame deletion of exons 2 to 7 spanning the extracellular ligand-binding domain that will produce a truncated, ligand-independent, constitutively phosphorylated receptor), which upon transplantation formed tumors with some oligodendroglial-like features (162). Some concerns with MMLV models have been the undefined cellular origin and the unknown contribution of retroviral insertional mutagenesis, the latter being applicable to retroviral models in general.
5.2 Transgenic Models of Oligodendroglioma
Transgenic mouse models have been extensively used to evaluate the significance of overexpression of oncogenes and targeted deletions of tumor suppressor genes in glioma. In addition, as exemplified above, many mouse lines carrying targeted deletions have been used in combination with retroviral models. When discussing transgenic models one needs to bear in mind that, contrary to viral models, the transgene analyzed will only be expressed as long as the promoter controlling its expression is active, which may explain why transgene models often are less efficient in generating tumors.
In 2003, Weiss et al. published a transgenic mouse model where a retroviral transforming allele of the EGF receptor, v-erbB, the viral homolog of the human EGFR that is lacking a large portion of the ligand-binding domain and has ligand-independent, constitutive tyrosine kinase activity much like the EGFRvIII, was expressed under the S100β promoter (7). Albeit S100β is expressed in many glial and neuronal cells of the CNS induced tumors were diagnosed as oligodendroglioma grade II. When cross-breeding the S100β-v-erbB mice with mice carrying targeted deletions of the p53 pathway, tumor development was accelerated and anaplastic oligodendroglioma developed. In a subsequent investigation, the cellular origin of tumors from the S100β-verbB model was deduced to be OPCs of the white matter (163). Similarly, Ding et al. described the generation of oligodendroglial-like tumors from combined transgenic expression of the EGFRvIII and V12Ha-Ras (RasB8), both under the control of the GFAP promoter (8). Also, transgenic expression of v-erbB in S100β expressing cells in rats produced oligodendrogliomas, anaplastic oligodendrogliomas, and glioblastomas (9, 164). Worth mentioning is that pCNPase-hEGFR transgenic mice did not develop any tumors (165). However, in this study the normal human EGFR was being used.
Data from human tumors suggests that aberrant PDGF signaling is more common than EGFR signaling in oligodendroglioma. However, transgenic models of PDGF expression have not been very potent in inducing gliomas. PDGF-B expressed under the control of the GFAP promoter had to be crossed with TP53 null mice to develop oligodendrogliomas and glioblastomas (166). Likewise, transgenic mice expressing PDGF-B from the nestin promoter did not produce any gliomas in the wild-type background (167). This could be due to inadequate expression levels and/or inaccurate spatiotemporal expression of PDGF-B for oligodendroglioma to develop.
5.3 Conclusions from Oligodendroglioma Modeling
Expression and mutation analyses of human oligodendroglioma have encouraged extensive studies on the role of PDGFR and EGFR signaling pathways using experimental glioma models. These studies have convincingly showed a causative role of these pathways in glioma development. Activation of PDGF signaling has been more potent than EGFR signaling in inducing experimental oligodendrogliomas in the viral models. When the EGFR mutant denoted EGFR* (168) was used intracranially in the RCAS/TV-A model, it resulted in the development of small glioma-like lesions, but only in combination with other genetic alterations (134). The higher oncogenic potency of PDGF activation may seem rational, since PDGFR overexpression is much more common in human tumors, and PDGFRα signaling is necessary and sufficient to maintain normal OPCs in a proliferative state (169–171). However, in transgenic models EGFR signaling appears to be more potent than PDGFR signaling, although definitive comparisons cannot be made since different promoters have been used. In normal development, EGFR signaling can stimulate oligodendrocyte production by expanding subventricular zone type-C progenitor cells (SVZ OPCs) that in turn will generate oligodendrocytes (172, 173). These cells were the putative target cells in the S100β-v-erbB mice (7). However, it cannot be excluded that PDGF signaling also contributed to tumor development in these mice since PDGFα-receptors were expressed in the tumor tissue. It has recently been shown that PDGFRα can be activated in a ligand-independent manner by activation of other receptor tyrosine kinases. These in turn will activate reactive oxygen species (ROS) that will phosphorylate PDGFRα (174), and in glioma cells it has been shown that ROS levels are clearly increased in response to EGFR activation (175).
The viral and transgenic models have made it possible to study the implication of cellular origin/differentiation status for glioma development. As discussed above, OPCs and/or oligodendrocytes have long been suggested to be the most likely cellular origin for oligodendroglioma. However, the results from the cell type-specific experimental models are proposing that experimental oligodendroglioma may develop from many different types of glial cells and that the oncogenic signaling pathways dictate the glioma subtype that develops. In the RCAS/TV-A model, PDGF signaling clearly favors oligodendroglioma development, while AKT + KRAS signaling will promote astrocytic tumor development (144, 176, 177). The origin of human oligodendroglioma remains undefined but the recent molecular-based stratification of human gliomas into different subclasses could in part reflect that they have originated from different cell types. Thus, taking the current knowledge into account we suggest that there is a common signaling pathway specific to oligodendroglioma that drives the phenotype of these tumors irrespective of cell of origin. This pathway appears to be related to normal OPC development and maintenance and tumors induced from neural/glial progenitor cells will acquire the histology of OPCs through a differentiation-like process, while tumors induced in astrocytes and CNPase positive OPCs will undergo a dedifferentiation-like process. Since these are not normal but neoplastic cells, this process may be more appropriately referred to as oncogene-induced differentiation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


