Chapter 10 Metabolic Acid-Base Disorders
Metabolic disturbances of acid-base balance are associated with many disease states, and identification of the acid-base disturbance may facilitate diagnosis of the underlying disease process. For example, observation of hypochloremic metabolic alkalosis on a serum biochemical profile of a vomiting dog may lead to recognition of gastrointestinal obstruction as the cause. The regulation of normal acid-base balance is considered in detail in Chapter 9.
Metabolic acidosis
Metabolic acidosis is characterized by a primary decrease in plasma HCO3− concentration, increased [H+], decreased pH, and a secondary, or adaptive, decrease in PCO2. In one study, metabolic acidosis was the most common acid-base disturbance in dogs and cats.61
Body buffer response to an acute acid load
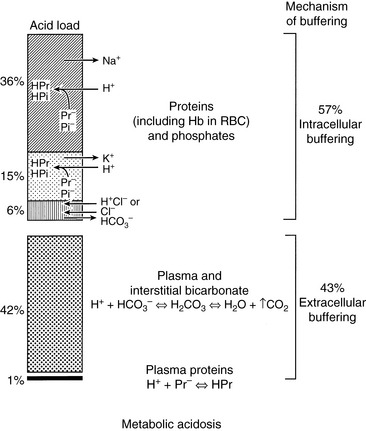
When HCl was infused acutely into nephrectomized dogs, approximately 40% of the acid was buffered by extracellular HCO3−, 10% by red cell buffers (primarily hemoglobin), and 50% by intracellular buffers of soft tissues and bone (primarily proteins and phosphates).223 In nonnephrectomized unanesthetized dogs infused intermittently with HCl, intracellular buffers contributed approximately 50% of the buffer response, regardless of the magnitude of the H+ load.210 Within a few minutes of an acute fixed acid load, administered H+ is buffered by HCO3− in plasma water. Plasma proteins and phosphates play a minor role in this acute response. Some of the administered acid enters red cells and is buffered by hemoglobin. The CO2 produced by the combination of the H+ with HCO3− ions is rapidly removed from the body by alveolar ventilation. Within 30 minutes, the acid load has been distributed to the interstitial fluid, where HCO3− again plays the dominant role in the acute buffer response. After several hours, H+ enters intracellular water in exchange for sodium and potassium ions. These hydrogen ions are buffered within cells by proteins and phosphates. In early studies,210,223 serum potassium concentration increased, but serum sodium concentration decreased after infusion of HCl. The relative roles of these buffers are depicted in Figure 10-1.
Respiratory response to an acute acid load
A fixed acid load increases [H+] and thereby stimulates peripheral and central chemoreceptors to increase alveolar ventilation. This effect begins within hours and is complete within 12 to 24 hours. In humans, there is an approximately 1.2-mm Hg reduction in PCO2 for each 1-mEq/L decrement in plasma HCO3− concentration to a minimum PCO2 of approximately 10 mm Hg.99,195 In dogs with uncomplicated metabolic acidosis induced by chronic feeding of HCl, the observed compensatory respiratory response is an approximately 0.7-mm Hg decrement in PCO2 per 1-mEq/L decrement in plasma HCO3− concentration.* In these studies, the smallest observed respiratory response was an approximately 0.5-mm Hg decrement in PCO2 per milliequivalents per liter decrement in plasma HCO3− concentration,3 and the largest response was a 1.1-mm Hg decrement in PCO2 per milliequivalents per liter decrement in plasma HCO3− concentration.66 Data are limited on the respiratory response of cats to metabolic acidosis, but there is some evidence that the cat fails to develop respiratory compensation to the same extent as observed in the dog in spontaneous236 and NH4Cl-induced metabolic acidosis.43,85,137,211,212
The classic explanation of the respiratory response to metabolic acidosis is that the increase in [H+] (decrease in pH) stimulates ventilation, and the resultant decrease in PCO2 returns the HCO3−/ PCO2 ratio and pH toward normal. This is true in acute metabolic acidosis, but the resultant secondary hypocapnia has been observed to decrease plasma HCO3− concentration further in chronic metabolic acidosis, presumably by reducing renal HCO3− reabsorption. This secondary hypocapnia contributes to 40% of the observed decrease in plasma HCO3− concentration during chronic HCl acidosis.147 Thus, chronic metabolic acidosis decreases plasma HCO3− concentration by two mechanisms: the effect of the administered HCl on body buffers and a reduction in renal HCO3− reabsorption that accompanies secondary hyperventilation. In this study, serum potassium concentration decreased during development of chronic HCl acidosis (contrary to what is typically described for acute metabolic acidosis caused by mineral acids), whereas serum sodium concentration was unchanged.147
Renal response to an acute acid load
The role of the kidneys is to excrete the fixed acid load imposed by the underlying disease process responsible for metabolic acidosis. The kidneys accomplish this task primarily by augmenting its excretion of NH4+. Titratable acidity changes little unless there is a change in the filtered load of phosphate. Chloride ions accompany the NH4+ into urine while HCO3− is regenerated and reabsorbed into extracellular fluid (ECF) to restore HCO3− that was titrated during the acute fixed acid load. Within 48 hours of a fixed acid load, approximately 25% of the added acid has been excreted in the urine, and the remainder is excreted during the next 4 days.232 The kidney can increase its NH4+ excretion as much as fivefold to tenfold during chronic metabolic acidosis.219,235,238 There is some evidence that cats do not adapt to metabolic acidosis by enhanced renal ammoniagenesis.137 The role of the kidneys in regulation of acid-base balance is discussed further in Chapter 9.
Clinical features of metabolic acidosis
Severe acidosis has serious detrimental effects on cardiovascular function, including decreased cardiac output, decreased arterial blood pressure, and decreased hepatic and renal blood flow.4 Myocardial contractility is decreased when blood pH falls below 7.20.161,180 Impaired contractility may result from a decrease in myocardial intracellular pH (pHi) and displacement of calcium ions from critical binding sites on contractile proteins. Acidosis may predispose the heart to ventricular arrhythmias or ventricular fibrillation. Acidosis has a direct arterial vasodilating effect that is offset by increased release of endogenous catecholamines. However, the inotropic response to catecholamines is impaired, and this may be associated with a reduction in the number of β-adrenergic receptors.151 Acidosis has a direct vasoconstrictive effect on the venous side of the circulation, which tends to centralize blood volume and predisposes to pulmonary congestion. Acidosis shifts the oxygen-hemoglobin dissociation curve to the right, thus enhancing O2 release from hemoglobin, but this effect is offset by a decrease in red cell 2,3-diphosphoglycerate, which develops after 6 to 8 hours of acidosis and shifts the curve back to the left.161
Acidemia produces insulin resistance that impairs peripheral uptake of glucose and inhibits anaerobic glycolysis by inhibiting phosphofructokinase.7 During severe acidosis, the liver may be converted from a consumer to a producer of lactate.144 Severe acidosis also impairs the ability of the brain to regulate its volume, leading to obtundation and coma. Acute mineral acidosis causes hyperkalemia by a transcellular shifting of potassium from intracellular fluid to ECF in exchange for hydrogen ions. This effect causes a very variable change in serum potassium concentration and is not observed with organic acidosis.6 Acute reduction in blood pH causes displacement of calcium ions from negatively charged binding sites (e.g., −COO− groups) on proteins (primarily albumin) as these sites become protonated, and an increase in ionized serum calcium concentration results. Chronic metabolic acidosis leads to release of buffer (mainly calcium carbonate) from bone, and osteodystrophy and hypercalciuria result.
Diagnosis of metabolic acidosis
Metabolic acidosis is associated with several different diseases and should be considered in any severely ill patient. Often, the diagnosis is first suspected by review of the electrolyte and total CO2 results on the patient’s biochemical profile. It is confirmed by blood gas analysis. The causes of metabolic acidosis may be divided into those associated with a normal anion gap (hyperchloremic metabolic acidosis) and those associated with an increased anion gap (normochloremic metabolic acidosis) (Box 10-1).
Box 10-1 Causes of Metabolic Acidosis
* Patients with diabetic ketoacidosis may have some component of hyperchloremic metabolic acidosis in conjunction with increased anion gap acidosis.7,9
† The metabolic acidosis early in renal failure may be hyperchloremic and later convert to typical increased anion gap acidosis.239
† Patients with hypoadrenocorticism typically have hypochloremia caused by impaired water excretion, absence of aldosterone, impaired renal function, and lactic acidosis. These factors prevent manifestation of hyperchloremia.
The anion gap represents the difference between the commonly measured plasma cations and the commonly measured anions. This concept is discussed in detail in Chapters 9 and 12. The normal electrolyte composition of canine plasma is compared with that in normal (hyperchloremic) and increased (normochloremic) anion gap metabolic acidosis in Figure 10-2. The anion gap concept is useful in the diagnostic approach to the patient with metabolic acidosis, but it must not be taken literally. In reality, electroneutrality is maintained, and there is no actual anion gap. Normally, the anion gap is made up of the net negative charge on sulfates, phosphates, plasma proteins, and organic anions (e.g., lactate, citrate). Recent studies have shown that in normal dogs and cats, a substantial portion of the anion gap arises from the negative charge on plasma proteins. The net protein charge of plasma at p. 7.40 was calculated to be 16.0 mEq/L in dogs,60 and this value was determined to be 13.7 mEq/L in cats.155 Factors other than metabolic acidosis also may affect the value of the anion gap, and these are discussed in Chapter 12.
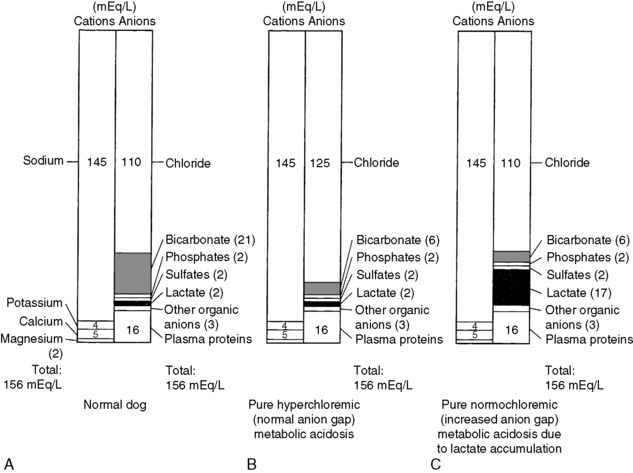
When the anion gap is calculated as [(Na+ + K+) − (Cl− + HCO3−)], normal values in dogs are in the range of 12 to 25 mEq/L.4,60,191,217 Values for the anion gap may be somewhat higher in cats (17 to 31 mEq/L) than in dogs (13 to 25 mEq/L) because of some unaccounted protein and phosphate charge.60,155 In other studies, the mean anion gap for normal cats (calculated as described above) was approximately 20 mEq/L.42,45,46 If the observed metabolic acidosis is characterized by a high anion gap, it is assumed to have arisen from an acid that does not contain chloride as its anion. Examples include some inorganic acids (e.g., phosphates, sulfates) or organic acids (e.g., lactate, ketoacids, salicylate, metabolites of ethylene glycol). In this setting, titration of body buffers by the acid results in accumulation of an anion other than chloride. If the observed metabolic acidosis is characterized by a normal anion gap, there is a reciprocal increase in the plasma chloride concentration to balance the decrease in plasma HCO3− concentration. In the following discussion, the causes of metabolic acidosis have been divided into those associated with a normal anion gap and those associated with an increased anion gap.
Disorders associated with a normal anion gap
Diarrhea
The concentration of HCO3− in intestinal fluid usually is higher than that of plasma, whereas its Cl− concentration is lower. This results from the addition of alkaline pancreatic and biliary secretions to luminal contents and from secretion of HCO3− in exchange for Cl− in the ileum (Fig. 10-3 and Table 10-1). In some diseases of the small intestine, increased delivery of ileal contents to the colon may overwhelm the considerable capacity of the colon for reabsorption of fluid and electrolytes. As a result, severe acute small bowel diarrhea may cause loss of HCO3− in excess of Cl− with resultant hyperchloremic metabolic acidosis. The acidosis is not purely hyperchloremic but rather is mixed if volume depletion and impaired tissue perfusion lead to lactic acid accumulation.
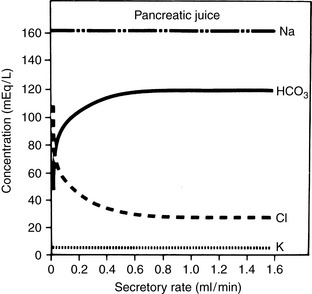
Table 10-1 Electrolyte Composition of Luminal Fluid at the End of Individual Segments of the Gastrointestinal Tract

In one study of 134 dogs with gastroenteritis caused by parvoviral infection, only 13% had low total CO2 concentrations.121 In another study of 17 dogs with parvoviral gastroenteritis, 59% had normal pH at presentation.108 In the animals with abnormal blood gas results, alkalemia (6 of 17) was more common than acidemia (1 of 17). The majority (64%) of the dogs in this study were presented for both vomiting and diarrhea. Hypochloremia is more common than hyperchloremia in parvoviral gastroenteritis.108,121 In another study consisting of 25 puppies with parvoviral enteritis, plasma concentrations of sodium, potassium, chloride, and bicarbonate were lower than those of control dogs; however, increases in serum L-lactate concentration were uncommon, and increases in serum D-lactate concentration were not observed.169 Most dogs in this study had mild compensated metabolic acidosis.
Renal Tubular Acidosis
Distal Renal Tubular Acidosis
In distal (classic or type 1) RTA, the urine cannot be maximally acidified because of impaired hydrogen ion secretion in the collecting ducts, and urine pH typically is above 6.0, despite moderately to markedly decreased plasma HCO3− concentration. Increased urine pH (>6.0) in the presence of acidosis is the hallmark of distal RTA. Urinary tract infection by a urease-positive organism (e.g., Proteus sp., Staphylococcus aureus) must be ruled out before considering distal RTA. Urinary net acid excretion is decreased, but bicarbonaturia usually is mild because urinary HCO3− concentration is only 1 to 3 mEq/L in the pH range of 6.0 to 6.5. Nephrolithiasis (usually calcium phosphate stones), nephrocalcinosis (resulting from alkaline urine pH and decreased urinary citrate concentration), bone demineralization (resulting from loss of bone buffer stores during chronic acidosis), and urinary potassium wasting with hypokalemia are features of distal RTA in human patients. Mutations in cytosolic carbonic anhydrase, the basolateral Cl−/HCO3− anion exchanger, and luminal H+-ATPase that affect function of the α-intercalated cells have been associated with inherited forms of distal renal tubular acidosis in humans.173 Urinary fractional excretion of HCO3− is normal (<5%) in distal RTA when plasma HCO3− concentration is increased to normal by alkali administration.
A diagnosis of distal RTA may be confirmed by an ammonium chloride tolerance test during which urine pH is monitored (using a pH meter) before and at hourly intervals for 5 hours after oral administration of 0.2 g/kg NH4Cl. Under such conditions, the urine pH of normal dogs decreased to a minimum value of 5.16 at 4 hours after administration of ammonium chloride.214 Dogs in this study also developed systemic acidosis (pH approximately 7.22 and HCO3− approximately 14 mEq/L at 2 hours after ammonium chloride administration). The amount of alkali required to correct the acidosis in human patients with distal RTA is variable but typically less than that required in proximal RTA. The required dosage of alkali in distal RTA may be as little as 1 mEq/kg/day (i.e., that required to offset daily endogenous acid production) or more than 2 to 4 mEq/kg/day. A combination of potassium and sodium citrate (depending on potassium balance) may be the preferred source of alkali.196
Proximal Renal Tubular Acidosis
In proximal (type 2) RTA, renal reabsorption of HCO3− is markedly reduced and urinary fractional excretion of HCO3− is increased (>15%) when plasma HCO3− concentration is increased to normal. Bicarbonaturia is absent and urine pH is appropriately low when metabolic acidosis is present and plasma HCO3− concentration is decreased because distal acidifying ability is intact. When plasma HCO3− concentration is decreased, the filtered load of HCO3− is reduced, and almost all of the filtered HCO3− is reabsorbed in the distal tubules, despite the presence of the proximal tubular defect. Thus, proximal RTA can be viewed as a “self-limited” disorder in which plasma HCO3− stabilizes at a lower than normal concentration after the filtered load falls sufficiently enough that distal HCO3− reabsorption can maintain plasma HCO3− at a new but lower steady-state concentration. Mutations in renal tubular transport proteins, such as the electrogenic basolateral Na+/ 3HCO3− cotransporter73 and one of the five forms of the luminal Na+/H+ antiporter, have been implicated in the pathogenesis of inherited forms of proximal renal tubular acidosis in humans.118
Multiple renal tubular reabsorptive defects resembling Fanconi syndrome have been reported in young basenji dogs.26–28,80 Clinical findings included polyuria, polydipsia, weight loss, dehydration, and weakness. Affected dogs had abnormal fractional reabsorption of glucose, bicarbonate, phosphate, sodium, potassium, and urate, and they had isolated cystinuria or generalized aminoaciduria. The renal tubular disorder in affected basenji dogs is thought to be the result of a metabolic or membrane defect affecting sodium movement or increased back leak or cell-to-lumen flux of amino acids. In one study, brush border membranes isolated from basenji dogs with Fanconi syndrome had decreased sodium-dependent glucose transport but no abnormality of cystine uptake despite the observed reabsorptive defect for cystine.157 Defective urinary concentrating ability leads to isosthenuria or hyposthenuria, and the GFR may be normal initially but decreased later in the course of the disease. Hypokalemia has also been observed late in the course of the disease.80 Death usually results from acute renal failure and papillary necrosis or acute pyelonephritis. A distinctive renal lesion is hyperchromatic karyomegaly of renal tubular cells.
Fanconi syndrome has been observed sporadically in other breeds81,143,156,182,213 and has been reported in association with administration of some drugs.16,28,160 In one case, Fanconi syndrome developed in association with primary hypoparathyroidism and resolved after treatment with calcium and calcitriol.88 Rickets in growing children and osteomalacia in adults are features of Fanconi syndrome in human patients that usually are not observed in affected dogs. However, congenital Fanconi syndrome and renal dysplasia were associated with histologic features of rickets in two Border terriers.64 The skeletal abnormalities in one of the affected dogs resolved after treatment with calcitriol and potassium phosphate. Transient Fanconi syndrome and proximal renal tubular acidosis also have been reported in a dog with high liver enzyme activities, and toxin exposure was considered as a possible explanation.115 Idiopathic transient renal tubular dysfunction also has been reported in a Labrador retriever126 and greyhound.1 Fanconi-like syndrome occurred in Australian dogs that had been fed dried chicken treats from China in 2007 and another product (not containing chicken and not from China) in 2009.87 Affected dogs had polyuria, polydipsia, glucosuria, acidosis, hypokalemia, hypophosphatemia, and azotemia. Most of them survived with conservative medical management. Finally, Fanconi syndrome has been reported in several dogs with copper storage hepatopathy, and tubular dysfunction resolved after copper chelation therapy.10,111
In one report, an 8-year-old female German shepherd had hyperchloremic metabolic acidosis, polyuria, polydipsia, isosthenuria, glucosuria with normal blood glucose concentration, and alkaline urine pH (7.46) after oral administration of NH4Cl.70 The metabolic acidosis was unresponsive to NaHCO3 administration at dosages up to 4 mEq/kg/day. This dog appeared to have distal (type 1) RTA and renal glucosuria. In another case of apparent distal RTA, a 5-year-old mixed breed dog was presented for evaluation of anorexia and was determined to have alkaline urine pH with hyperchloremic metabolic acidosis.191 In another report, an 8-year-old female German shepherd was presented for polyuria, polydipsia, weight loss, and lethargy.24 It had a normal GFR, metabolic acidosis, hyposthenuria, and intermittent glucosuria. Fractional reabsorption of sodium, glucose, and HCO3− was decreased, but reabsorption of chloride, phosphate, potassium, urate, and amino acids was normal. The dog gained weight, and its clinical signs were reversed after treatment with NaHCO3 at approximately 10 mEq/kg/ day. This dog appeared to have proximal (type 2) RTA.
Distal RTA has been reported in two cats with pyelonephritis caused by Escherichia coli.77,236 Clinical signs included polyuria, polydipsia, anorexia, lethargy, enlarged kidneys, and isosthenuria. In one cat, urine pH was 5.0 at the time pyelonephritis was first diagnosed, but distal RTA was documented at a later time by the presence of hyperchloremic metabolic acidosis, alkaline urine pH, and failure to lower urine pH after oral administration of NH4Cl.77 Findings were similar for the other cat, but hyperphosphaturia and persistent hypokalemia also were detected.236 Distal RTA and hepatic lipidosis were reported in another cat without urinary tract infection29 and in a cat with concurrent hyperaldosteronism and severe hypokalemia.228 Distal renal tubular acidosis also has been reported in association with immune-mediated hemolytic anemia in three dogs.215 Distal renal tubular acidosis is associated with some immune-mediated diseases in human patients, but not specifically immune-mediated hemolytic anemia. The clinical features of proximal (type 2) and distal (type 1) RTA are summarized in Table 10-2.
Table 10-2 Clinical Features of Proximal and Distal Renal Tubular Acidosis
| Clinical Feature | Proximal RTA | Distal RTA |
|---|---|---|
| Hypercalciuria | Yes | Yes |
| Hyperphosphaturia | Yes | Yes |
| Urinary citrate | Normal | Decreased |
| Bone disease | Less severe | More severe |
| Nephrocalcinosis | No | Yes |
| Nephrolithiasis | No | Yes (calcium phosphate) |
| Hypokalemia | Mild | Mild to severe |
| Potassium wasting | Worsened by NaHCO3 | Improved by NaHCO3 |
| Alkali required for treatment | >10 mEq/kg/day | <3 mEq/kg/day |
| Other defects of proximal tubular function* | Yes | No |
| Reduction in plasma HCO3− | Moderate | Variable (can be severe) |
| FeHCO3− at normal plasma HCO3− concentration | >15% | <5% |
| Urine pH during acidemia | <5.5 | >6.0 |
| Urine pH after Nh4cl | <5.5 | >6.0 |
Fe, fractional excretion.
* Decreased fractional reabsorption of sodium, potassium, phosphate, urate, glucose, and amino acids.
Hyporeninemic hypoaldosteronism, characterized by hyperkalemia with decreased plasma renin and aldosterone concentrations, occurs in some human patients, notably those with diabetes mellitus who also have mild to moderate renal insufficiency.65 The hyperchloremic metabolic acidosis observed in these patients has been called Type 4 RTA. This syndrome has not been characterized in veterinary medicine but should be considered in dogs and cats with hyperkalemia and mild to moderate hyperchloremic metabolic acidosis after hypoadrenocorticism has been ruled out by an adrenocorticotropic hormone (ACTH) response test. The diagnosis may be established by finding an inappropriately decreased plasma aldosterone concentration in the presence of hyperkalemia.
Carbonic Anhydrase Inhibitors
Carbonic anhydrase inhibitors, such as acetazolamide, decrease proximal tubular reabsorption of HCO3− in the kidneys by noncompetitive inhibition of luminal and cellular carbonic anhydrase. Hypokalemia is caused by increased sodium delivery to the distal nephron and its reabsorption there in exchange for potassium. As hyperchloremic metabolic acidosis develops, the filtered load of HCO3− decreases and the effect of carbonic anhydrase inhibitors on HCO3− reabsorption is limited. Acetazolamide given at 7 to 10 mg/kg three times daily causes self-limited hyperchloremic metabolic acidosis, mild to moderate hypokalemia, and mild hypocalcemia in dogs.107,201 The effects of acetazolamide were greatest after 3 days of administration, and blood chemistry results stabilized after 5 days of administration.201 Acetazolamide is used most commonly in small animal practice for the treatment of glaucoma.
Ammonium Chloride
Administration of NH4Cl is equivalent to administration of HCl because the NH4+ is converted in the liver to urea and H+. Ammonium chloride has been used commonly as a urinary acidifier in dogs and cats. A study of cats receiving 800 mg of NH4Cl per day as a powder or tablet showed that venous blood pH and HCO3− concentrations were decreased to values at the lower end of the normal range.211 A combination product supplying 580 mg each of NH4Cl and D,L-methionine had a more notable effect on venous blood pH and HCO3− concentrations than that observed with 800 mg of NH4Cl alone, but results were still within the reported normal range.212 In another study of cats, NH4Cl at 300 mg/kg/day did not significantly alter venous blood pH, PCO2, or HCO3− concentration, but 400 mg/kg/day significantly decreased blood HCO3− concentration during the course of the study.85 Ammonium chloride at a dosage of 535 mg/kg/day administered to dogs over 6 days caused hyperchloremic metabolic acidosis and was associated with hypokalemia, presumably related to increased aldosterone secretion.150 In another study of dogs, NH4Cl at 200 mg/kg/day reduced urine pH to approximately 5.0 and produced mild metabolic acidosis without change in serum potassium concentration.208
In young, growing and adult dogs, the addition of NH4Cl to the diet leads to demineralization of bone.34,125 Chronic acid feeding has also been reported to affect bone metabolism in cats. Diets containing 3% NH4Cl slowed growth of young cats, decreased blood pH and HCO3− concentrations, and lowered urine pH. Urinary calcium excretion increased in these cats, and bone demineralization was observed on histologic examination of caudal vertebrae.32 Adult cats fed 1.5% NH4Cl for 6 months developed hyperchloremic metabolic acidosis and negative balance for calcium and potassium,43 but no significant changes in trabecular bone remodeling or bone mineral density were found.44 In one study, administration of NH4Cl to cats fed a potassium-restricted diet resulted in hypokalemia, possibly by reducing gastrointestinal absorption of potassium.76 Results of these studies indicate that NH4Cl should be used with caution and blood gases should be monitored during therapy.
Posthypocapnic Metabolic Acidosis
During compensation for chronic respiratory alkalosis, renal net acid excretion decreases with consequent reduction in plasma HCO3− and increase in plasma Cl− concentrations. When the stimulus for hyperventilation is removed and PCO2 increases, pH decreases because it requires 1 to 3 days for the kidneys to increase net acid excretion and to increase plasma HCO3− concentration. Until this occurs, a state of “posthypocapnic” metabolic acidosis exists. Recovery is spontaneous as long as sodium and phosphate are available in the diet to allow the appropriate increase in renal net acid excretion.90
Hypoadrenocorticism
Aldosterone increases renal tubular lumen negativity by enhancing sodium reabsorption in the collecting duct and secondarily increases hydrogen ion secretion. It also directly stimulates H+ secretion by increasing the activity of the luminal H+-ATPase pump in the medullary collecting duct. These effects allow urinary excretion of H+ and K+ when distal delivery of sodium is decreased. Deficiency of aldosterone in hypoadrenocorticism results in metabolic acidosis and hyperkalemia. Metabolic acidosis of variable severity is common in dogs with hypoadrenocorticism.159,190 In one study, low total CO2 concentration suggesting the presence of metabolic acidosis was found in 81 of 200 (41%) dogs with hypoadrenocorticism.190 In a study of 10 cats with hypoadrenocorticism, 3 were reported to have decreased serum total CO2 concentrations.189 Treatment of hypoadrenocorticism includes volume expansion with 0.9% NaCl and replacement of deficient mineralocorticoids and glucocorticoids.
Disorders associated with an increased anion gap
Ethylene Glycol Ingestion
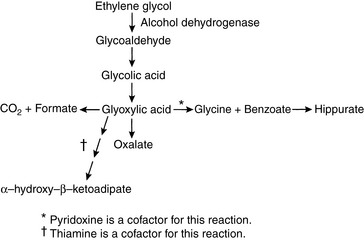
Ethylene glycol (EG) is an organic solvent (molecular mass, 62 Da) used in commercial antifreeze solutions. Ingestion of antifreeze by dogs and cats is a common cause of oliguric acute renal failure in small animal practice, and mortality exceeds 80% in affected animals.57,95,227 EG itself is not toxic, but it is converted in the liver to several metabolites that cause severe metabolic acidosis and acute renal failure (Fig. 10-4). It is rapidly absorbed from the gastrointestinal tract and is undetectable in plasma of dogs 48 hours after administration.175,205
Pathophysiology
EG is first metabolized in the liver to glycoaldehyde by alcohol dehydrogenase. Glycoaldehyde uncouples oxidative phosphorylation and may contribute to neurologic signs observed early in the course of intoxication. Subsequent steps in metabolism produce glycolic and glyoxylic acids. Glycolic acid is primarily responsible for the severe metabolic acidosis that occurs in animals poisoned by EG.50 Renal tubular injury results from glycoaldehyde, glycolic acid, and glyoxylic acids, and calcium oxalate crystals are deposited within renal tubules. The observation of these birefringent crystals in the presence of acute tubular nephrosis confirms the diagnosis of EG intoxication.
Vomiting, polydipsia, and polyuria may occur soon after ingestion of EG, but the owners of poisoned animals often do not detect these signs. Within 12 hours of ingestion, neurologic signs (e.g., lethargy, ataxia, stupor, seizures, coma) may develop. Cardiac and pulmonary manifestations (e.g., tachypnea, tachycardia) occur 12 to 24 hours after ingestion but rarely are detected in clinical cases. Oxalate crystals may be detected in the urine as early as 3 to 6 hours after ingestion of EG.68,69 Renal failure occurs in dogs as early as 24 to 48 hours after ingestion and is manifested by anorexia, lethargy, vomiting, and oliguria or anuria.97 In cats, azotemia may develop within 12 to 24 hours after ingestion of EG.68 Unfortunately, most dogs and cats with EG poisoning are presented for veterinary attention after renal failure has already developed.
A severe normochloremic (i.e., high anion gap) metabolic acidosis occurs within 3 hours of EG ingestion and persists for at least 24 hours.68,69,97,227 Serum hyperosmolality and osmolal gap peak 1 to 6 hours after ingestion and persist for 12 to 24 hours,68,69,97 but the osmolal gap may be normal in animals presented later in the course of the disease.227 Activated charcoal preparations containing propylene glycol and glycerol can increase osmolality and osmolal gap, and potentially complicate the diagnosis of EG ingestion. Measured serum osmolality peaked at 4 hours (353 mOsm/kg), osmolal gap at 6 hours (52 mOsm/kg), and serum lactate concentration at 4 hours (4.5 mmol/L) after administration of 4 g/kg of an activated charcoal preparation containing propylene glycol and glycerol.33 Results returned to baseline 24 hours after administration of the activated charcoal preparation.
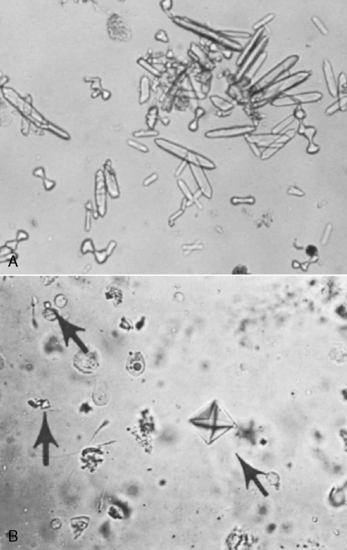
Calcium oxalate dihydrate crystals (“Maltese cross” or “envelope” forms) may be observed in the urine, but calcium oxalate monohydrate crystals (“picket fence” or “dumbbell” forms) are observed more commonly. Calcium oxalate dihydrate crystals occasionally are found in the urine of normal dogs and cats, whereas calcium oxalate monohydrate crystals rarely are seen except in animals that have ingested EG (Fig. 10-5).68,227 Crystals previously referred to as hippurates actually are calcium oxalate monohydrate crystals.134,226 Other laboratory findings include azotemia, isosthenuria, hypocalcemia, hyperphosphatemia, and hyperglycemia.227 Hyperphosphatemia observed very early in the course of EG intoxication (3 to 12 hours after ingestion) probably is the result of the high phosphorus content of rust-retardant antifreeze preparations.57,69 Hyperechogenicity of the renal cortex is observed on renal ultrasonography as early as 5 hours after ingestion of EG.2
Treatment
The response to treatment depends on the amount of EG ingested and the amount of time that elapses before treatment. In early studies, dogs that ingested less than 10 mL/kg EG were saved if treated within 2 to 4 hours of ingestion,17,175,205 and cats survived up to 6 mL/kg EG if treated within 4 hours.187 Treatment consists of inducing vomiting with apomorphine or performing gastric lavage with activated charcoal if ingestion has been recent (<8 hours before presentation). Severe hypocalcemia is corrected with calcium gluconate, and NaHCO3 is administered to combat metabolic acidosis. A NaHCO3 dosage of 1 to 2 mEq/kg may be used empirically. Calcium gluconate and NaHCO3 must not be given simultaneously because calcium carbonate crystals form, and the solution becomes turbid. Attempts to stimulate urine production with furosemide (2 to 4 mg/kg) or mannitol (1 g/kg) usually are futile.
Alcohol dehydrogenase has greater affinity for ethanol than EG. For this reason, 20% ethanol has been administered intravenously to affected dogs at a dosage of 5.5 mL/kg every 4 hours for five treatments and then every 6 hours for four additional treatments.96 Cats are treated with 20% ethanol at a dosage of 5 mL/kg every 6 hours for five treatments and then every 8 hours for four additional treatments. This treatment is unlikely to be of benefit if more than 12 to 24 hours have elapsed since ingestion of EG. Fomepizole (4-methylpyrazole) is a pharmacologic inhibitor of alcohol dehydrogenase that can be used to treat dogs with EG toxicosis.67,69 In dogs, it is superior to ethanol because it does not cause central nervous system (CNS) depression, but it must be administered within 8 hours of EG ingestion. The dosage of fomepizole used in dogs with EG intoxication is 20 mg/kg intravenously, followed by 15 mg/kg intravenously at 12 and 24 hours and 5 mg/kg intravenously at 36 hours.57,67,69 Unfortunately, fomepizole was not efficacious in EG-intoxicated cats unless administered at the same time as the EG was consumed.68 A study to investigate the difference in efficacy of fomepizole between dogs and cats found that the percentage inhibition of canine and feline alcohol dehydrogenase was similar when the concentration of fomepizole applied to feline liver homogenates was 6 times higher than that applied to canine liver homogenates.58 When cats that received lethal doses of EG were treated within 3 hours of ingestion using 125 mg/kg fomepizole followed by 31 mg/kg at 12, 24, and 36 hours, 5 of 6 survived.59 One cat developed acute renal failure but recovered. Cats treated with this high dosage of fomepizole developed mild sedation, but no biochemical evidence of toxicity was identified.
Thiamine promotes conversion of glyoxylate to glycine, and pyridoxine promotes conversion of glyoxylate to α-hydroxy-β-ketoadipate (see Fig. 10-4). These vitamins may be administered to promote alternative pathways of glyoxylate metabolism, but efficacy has not been demonstrated for such treatment. In one study, all nonazotemic dogs treated with fomepizole within 2 to 8.5 hours after EG ingestion survived, whereas only 1 of 21 azotemic dogs treated 8.5 to 38 hours after ingestion survived.57
Peritoneal dialysis or hemodialysis is necessary if the animal has anuric or oliguric renal failure at the time of presentation. Early dialysis may also be helpful to remove toxic intermediate metabolites. Despite dialysis, affected dogs may progress to end-stage renal disease and become dependent on dialysis. The prognosis for survival in adult dogs and cats with anuric or oliguric acute renal failure caused by EG intoxication is unfortunately very poor.57,227
Salicylate Intoxication
Aspirin (acetylsalicylic acid) is hydrolyzed to salicylic acid (pK’a = 3.0) in the liver. Salicylate intoxication is uncommon in small animal practice and is an example of a mixed acid-base disturbance characterized by metabolic acidosis and respiratory alkalosis. Salicylate intoxication in anesthetized, spontaneously breathing dogs resulted in a mixed respiratory alkalosis and metabolic acidosis.218 The stimulation of ventilation is caused by a direct effect of salicylate on the medullary respiratory center. Salicylate also uncouples oxidative phosphorylation in mitochondria, and the associated disturbances in carbohydrate metabolism lead to metabolic acidosis characterized by an increased anion gap associated with accumulation of lactic acid, ketoacids, and other organic acids. Salicylate usually makes a minor contribution to the observed increase in unmeasured anions.
Metaldehyde Intoxication
Metaldehyde is a tetramer of acetaldehyde used as a snail and slug bait that can cause seizures and hyperthermia in dogs that ingest it.241 It is hydrolyzed to acetaldehyde in the stomach, which then is absorbed and metabolized to acetic acid (pK’a = 4.75). Acidemia was present in 6 of 11 intoxicated dogs in which arterial blood gas analysis was performed. Three dogs had metabolic acidosis and three had mixed acid-base disturbances that were not further characterized. Conversion of acetaldehyde to acetic acid could explain development of metabolic acidosis, and ventilatory disturbances associated with generalized seizures (either respiratory alkalosis or acidosis) could have contributed to development of mixed acid-base disorders. With supportive care, blood gas abnormalities resolve within 24 to 48 hours.
Diabetic Ketoacidosis
Pathophysiology
Overproduction of acetoacetic acid (pK’a = 3.58) and β-hydroxybutyrate (pK’a = 4.70) by the liver occurs in diabetes mellitus because of a deficiency of insulin and relative excess of glucagon. An increase in glucagon and a decrease in insulin shift the liver from its normal role in esterification of fatty acids into triglycerides to β-oxidation of fatty acids into ketoacids. At the normal pH of ECF (7.40), these organic acids are completely dissociated, and the hydrogen ions that are released titrate HCO3− and other body buffers. Acetone is formed by the nonenzymatic decarboxylation of acetoacetate and does not contribute additional fixed acid. The pathophysiology and treatment of diabetic ketoacidosis are discussed in detail in Chapter 20.
Metabolic acidosis is common in dogs and cats with diabetic ketoacidosis. In one series, mean plasma HCO3− concentration in 72 dogs with diabetic ketoacidosis was approximately 11 mEq/L at the time of diagnosis with a range of 4 to 20 mEq/L, whereas the mean HCO3− concentration in 20 affected cats was 13 mEq/L with a range of 8 to 22 mEq/L.83 In an early study of dogs with diabetes mellitus, mean plasma HCO3− concentration was 13.7 mEq/L in eight survivors (range, 9.3 to 21.0 mEq/L) and 18.1 mEq/L in five nonsurvivors (range, 13.4 to 30.2 mEq/L).138 In another study of dogs with diabetic ketoacidosis, mean arterial pH and HCO3− concentration were 7.201 (range, 6.986 to 7.395) and 11.1 mEq/L (range, 4.1 to 19.7 mEq/L) before treatment and 7.407 ± 0.053 and 18.2 ± 0.7 mEq/L 24 hours after treatment.142 Only three dogs (those with pH <7.1) received sodium bicarbonate treatment. Metabolic acidosis with median pH of 7.14 (range, 7.04 to 7.24) and HCO3− concentration of 10 mEq/L (range, 6 to 15 mEq/L) was found in 25 of 33 cats evaluated by venous blood gas analysis in a survey of cats with diabetic ketoacidosis.31 Cats with HCO3− concentrations below 14 mEq/L received bicarbonate supplementation of their fluids. In another series of diabetic cats, median total CO2 was 13 mEq/L in ketoacidotic cats and 15 mEq/L in nonketoacidotic cats.63 In a study of 116 dogs with diabetes mellitus, 43 (37%) had diabetic ketoacidosis with median venous blood pH of 7.228 (range, 6.979 to 7.374) and median bicarbonate concentration of 10.1 mEq/L (range, 4.0 to 19.3 mEq/L).78 In a study of 127 dogs with ketoacidosis, acid-base status at presentation had substantial impact on outcome.117 Nonsurvivors had lower venous pH and larger base deficits, and for each unit improvement in base deficit there was a 9% increase in likelihood of discharge from the hospital.
The nitroprusside reagent (e.g., Acetest, Bayer, Tarrytown, N.Y.) detects only ketone (−C=O) groups (e.g., acetoacetate, acetone). The concentration of β-hydroxybutyrate typically exceeds that of acetoacetate in uncontrolled diabetic ketoacidosis, and the dipstick reaction underestimates the degree of ketonuria. This problem can be overcome by adding a few drops of hydrogen peroxide to urine, which nonenzymatically converts β-hydroxybutyrate to acetoacetate.171 When insulin is administered and metabolism of ketones proceeds, there is a shift toward acetoacetate, and the dipstick reaction transiently becomes more strongly positive. This possibility should be recognized by the clinician and should not cause concern. In a study of 116 diabetic dogs (of which 88 had not previously received insulin), all ketotic and ketoacidotic dogs and 21 of 32 (66%) “nonketotic” dogs (i.e., negative urine dipstick test for ketones) had abnormally high serum β-hydroxybutyrate concentrations (>0.15 mmol/L) at presentation.78 Although not as readily available, measurement of plasma ß-hydroxybutyrate concentrations is more valuable than use of dipstick tests in the characterization of ketonemia in diabetic dogs and cats.74,242,243 The increase in unmeasured anions (as reflected in the anion gap) gives a rough estimate of the concentration of ketoanions in serum. However, this estimate is inaccurate if lactic acidosis develops because lactate also is an unmeasured anion. In one study of diabetic dogs, however, acidosis was correlated primarily with serum ketone concentration, and not with serum lactate concentration.79
To some extent, the anions of these ketoacids are excreted in urine along with sodium and potassium for electroneutrality. These organic anions are lost from the body and cannot be metabolized to HCO3− after correction of diabetic ketoacidosis with insulin therapy. Their loss thus contributes to depletion of body buffer and cation stores. Osmotic diuresis is induced by hyperglycemia and also contributes to the whole-body cation deficit. The extent of impairment in renal function may determine whether patients with diabetic ketoacidosis have an increased anion gap metabolic acidosis or hyperchloremic metabolic acidosis at the time of presentation. Patients with severe volume depletion have an increased anion gap because of retention of ketoanions, whereas those without volume depletion have hyperchloremia as a result of increased urinary excretion of the sodium and potassium salts of ketoanions and retention of chloride.5,9
Treatment
The best treatment for the acidosis of uncontrolled diabetes mellitus is fluid therapy and insulin. Insulin administration allows glucose use by skeletal muscle and adipose tissue, decreases hepatic glucose production, prevents lipolysis and ketogenesis, and permits peripheral metabolism of ketoacids. Several regimens for administration of insulin to ketoacidotic dogs and cats have been described.84 The particular protocol of insulin administration is probably less crucial to the ultimate outcome than the individualized care provided by the veterinarian during management of the diabetic animal.
Several factors may contribute to a delay in the repair of the HCO3− deficit in patients with diabetic ketoacidosis.100 Ketoacid anions that have been excreted in the urine are lost to the body and cannot be metabolized to HCO3−. After treatment with fluids and insulin, recovery may be faster in patients with a high anion gap because the retained ketoanions are metabolized, yielding HCO3−.7,9 Thus, withholding alkali may be more rational for diabetic patients with high anion gap metabolic acidosis than for those with hyperchloremic metabolic acidosis. Dilutional acidosis may occur if ECF volume (ECFV) is expanded with alkali-free solutions such as 0.9% saline. If hyperventilation persists, it may impair renal reabsorption of HCO3−, and renal acid excretion may require several days to become fully augmented.
The use of NaHCO3 to treat diabetic ketoacidosis is highly controversial, and clear benefits of its use have not been demonstrated in human patients. For example, there was no difference in recovery (based on rate of decrease of blood glucose and ketone concentrations and rate of increase of blood or cerebrospinal fluid [CSF] pH or HCO3− concentration) when NaHCO3 was or was not administered to human patients with diabetic ketoacidosis who presented with blood pH values in the range of 6.90 to 7.14.166 In another study, treatment with NaHCO3 delayed resolution of ketosis in diabetic ketoacidosis.178
There are several theoretical arguments against the use of NaHCO3 in diabetic ketoacidosis. Acidosis in the CNS may develop after NaHCO3 administration. The blood-brain barrier is permeable to CO2 but less permeable to the charged HCO3− ion. If NaHCO3 is administered, pH increases in ECF as the HCO3−/ PCO2 ratio increases, and compensatory hyperventilation decreases somewhat. As a result, PCO2 increases and CO2 diffuses into the CNS. However, bicarbonate diffusion into CNS lags behind that of CO2. During this time, the HCO3− PCO2 ratio and pH in the CNS may decrease. This has been referred to as paradoxical CNS acidosis.192 The frequency of occurrence of this complication and its clinical significance are uncertain.135
Uremic Acidosis
Pathophysiology
The metabolic acidosis of chronic renal failure is usually mild to moderate in severity (plasma HCO3− concentration, 12 to 15 mEq/L) and may be hyperchloremic early in the course of the disease process.239 Later in the course of the disease, the anion gap increases because of retention of phosphates, sulfates, and organic anions. Acid-base status is usually well preserved in chronic renal failure until GFR decreases to 10% to 20% of normal. In retrospective studies of small animal patients with chronic renal failure, plasma HCO3− concentrations were less than 16 mEq/L in 40% of dogs with chronic renal failure caused by amyloidosis72 and less than 15 mEq/L in 63% of cats with chronic renal failure of various causes.71 A high anion gap was observed in 43% of affected dogs (>25 mEq/L) and in 19% of affected cats (>35 mEq/L) in these studies. In acute renal failure, there has been insufficient time for the kidneys to adapt to the disease state, and the metabolic acidosis of acute renal failure is usually more severe than that observed in chronic renal failure. Complications such as sepsis and marked tissue catabolism may contribute to the severity of metabolic acidosis in acute renal failure.
Delivery of HCO3− from the proximal tubules to the distal nephron is increased in chronic renal failure.235 In dogs with experimentally induced unilateral renal disease, renal HCO3− reabsorption was not different in the diseased and control kidneys, but bicarbonaturia developed when the normal kidney was removed, and the contralateral diseased kidney was forced to function in a uremic environment.165 The osmotic diuresis characteristic of uremia may thus contribute to the increased delivery of HCO3− to the distal tubules. Increased parathyroid hormone concentration as a result of renal secondary hyperparathyroidism does not seem to have important adverse effects on HCO3− reabsorption in experimentally induced renal disease in dogs.12,206,207 The ability to lower urine pH maximally is preserved in chronic renal failure.
The main method by which the diseased kidney responds to chronic retention of fixed acid is by enhanced renal ammoniagenesis. Total ammonium excretion decreases during progressive chronic renal disease, but ammonium excretion is observed to be markedly increased when expressed per 100 mL GFR or per remnant nephron.75,202 On a per nephron basis, the diseased kidney can increase its ammonium excretion threefold to fivefold.219,235,238 This adaptive mechanism seems to be fully expended when the GFR decreases to less than 20% of normal. At this point, the diseased kidneys can no longer effectively cope with the daily fixed acid load, and a new steady state is established at a lower than normal plasma HCO3− concentration. The relatively mild decrease in plasma HCO3− concentration that is observed in chronic renal failure has been attributed to the contribution of the large reservoir of buffer (e.g., calcium carbonate) in bone. However, the capacity of the skeleton to buffer the amount of acid that accumulates in long-standing chronic renal failure has been questioned.177 The decrease in total ammonium excretion that occurs in chronic renal failure may be counterbalanced by decreased urinary excretion of organic anions (e.g., citrate, lactate, pyruvate, ketoanions).56 Metabolism of these retained organic anions would result in a net gain of HCO3− that would offset the decreased excretion of H+ in the form of NH4+.
The amount of phosphate buffer available in urine in chronic renal failure is relatively fixed and likely to be at its maximum because of hyperphosphatemia and the effects of increased plasma parathyroid hormone concentration.202,219 Furthermore, phosphorus binders and dietary phosphorus restriction are commonly used to treat chronic renal failure and may limit the amount of phosphate that can contribute to titratable acidity. When expressed on a per nephron basis, however, titratable acidity is increased in chronic renal failure.164
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


