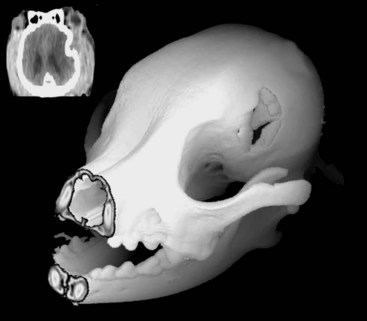
Chapter 35 Severe brain injury in dogs and cats is an unfortunately frequent occurrence and is often associated with a guarded to poor prognosis, even with aggressive management. Much of the information regarding therapy for these patients is adapted from the human literature, although several veterinary-specific studies are available.15,20 In addition to the challenge of choosing and administering appropriate medical therapy for acute severe brain injury, the surgeon is often faced with the decision regarding whether or not surgical intervention is indicated for a particular patient. Although some differences in opinion have been expressed over what constitutes “appropriate” medical therapy for severe brain injury in veterinary and human patients, general agreement has been reached on most of the available treatments. Less agreement is seen among clinicians with respect to surgical intervention for severe brain injury, particularly in scenarios in which surgical therapy is being used as a decompressive maneuver in the absence of intracranial hemorrhage.13 This chapter will review the pathophysiology and treatment principles associated with severe brain injury in dogs and cats. Because of the lack of published information in the veterinary literature regarding surgical management of severe brain injury, the discussion on this aspect of management will be based on a combination of the human literature and the authors’ clinical experience. Primary brain injury refers to the physical disruption of intracranial structures that occurs immediately at the time of the traumatic event. Such injury includes direct damage to brain parenchyma, such as contusions, lacerations, and diffuse axonal injury. Damage to blood vessels may result in intracranial hemorrhage and vasogenic edema. Skull fractures can contribute to continued trauma to the brain parenchyma and blood vessels, especially if they are unstable (Figure 35-1). The extent of primary brain injury is a function of the force of impact. Acceleratory and deceleratory forces of both the impacting object(s) and the intracranial contents will affect overall tissue damage. Direct parenchymal damage associated with primary brain injury is generally beyond the control of the clinician. However, stabilization of skull fractures and evacuation of intracranial hemorrhage may decrease the morbidity associated with these primary injuries. In addition to continued hemorrhage and edema, the damage caused by the primary brain injury activates a number of interrelated biochemical pathways that act in concert to perpetuate further brain tissue damage and subsequent increases in intracranial pressure. These processes constitute secondary brain injury. Adenosine triphosphate (ATP) depletion disrupts the maintenance of cellular ionic homeostasis. Sudden, uncontrolled intracellular influx of sodium (Na+) and calcium (Ca2+) occurs. Cellular swelling (cytotoxic edema) and depolarization result. Uncontrolled depolarization leads to the release of large amounts of glutamate, an excitatory neurotransmitter, into the extracellular environment. Glutamate causes further increases in intracellular Ca2+ levels. Elevated Ca2+ levels activate a number of tissue-damaging pathways, including the arachidonic acid cascade (phospholipase A2 activation) and the xanthine oxidase (free radical producing) pathway. Iron (Fe2+) is a vital co-factor in the xanthine oxidase pathway, and free radical species generated via the Fenton reaction (e.g., hydroxyl and superoxide radicals) are preferentially damaging to cell membranes containing high levels of polyunsaturated fats and cholesterol. Brain tissue is rich in both Fe2+ and membranes with high levels of polyunsaturated fats and cholesterol. Intraparenchymal hemorrhage also increases the amount of Fe2+ available for perpetuation of oxidative damage. Free radical species are thus particularly damaging to neuronal membranes and probably play a major role in secondary brain injury. Their production is also induced by ischemia, arachidonic acid metabolites, catecholamine oxidation, and activated neutrophils. Other secondary autolytic processes induced after severe head trauma include the complement, kinin, and coagulation/fibrinolytic cascades. Elevated levels of nitric oxide and various cytokines (e.g., tumor necrosis factor, interleukins) also contribute to parenchymal injury in the damaged brain. Most of the mediators of tissue damage produced by these various reactions perpetuate their own continued production, as well as the production of other mediators. The maintenance of an ischemic environment perpetuates the previously mentioned processes and also leads to the accumulation of lactic acid (via anaerobic glycolysis). Lactic acid accumulation causes further damage to brain tissue. Hypotension and hypoxemia, extracranial conditions that are common in the traumatized patient, can worsen brain ischemia and thereby enhance the events responsible for secondary brain injury. The end result of these secondary processes is increased intracranial pressure. Unlike in primary brain injury, the clinician has some control over secondary brain injury. where MABP = mean arterial blood pressure The normal contents of the cranial cavity include brain parenchyma, blood, and cerebrospinal fluid. In the normal animal, these components exist in equilibrium with each other, and intracranial pressure remains within normal limits. Between the mean arterial blood pressure extremes of 50 and 150 mm Hg, intracranial pressure remains constant. This phenomenon is called pressure autoregulation. Pressure autoregulation serves to link systemic blood pressure changes to brain vasculature tone. If mean arterial blood pressure rises, vasoconstriction occurs in the brain; if mean arterial blood pressure falls, vasodilation occurs in the brain (see Figure 29-2). In the normal animal, the former scenario prevents intracranial pressure from rising by decreasing cerebral blood flow, and in the latter, intracranial pressure is prevented from falling by increasing cerebral blood flow. Chemical autoregulation refers to the direct responsiveness of brain vasculature to the partial pressure of carbon dioxide in arterial blood (PaCO2); elevated PaCO2 levels cause cerebral vasodilation, whereas decreased PaCO2 levels cause cerebral vasoconstriction. Both forms of autoregulation often remain intact in people with severe head injury, but pressure autoregulation may be compromised in approximately 30% of patients. In some of these individuals, the lower mean arterial blood pressure extreme may become “reset” to a higher value, resulting in significantly decreased blood flow to the brain with even mild systemic hypotension. With severe head trauma, both intracranial hemorrhage and edema can add to the volume of the intracranial compartment. Because of the inexpansile nature of the skull, one or more components of the cranial cavity must accommodate for the increased volume, or increased intracranial pressure will result. This accommodation or volume buffering is accomplished by fluid shifts in the brain vasculature and cerebrospinal fluid pathways and is referred to as intracranial compliance. Compliance is expressed as the change in volume per unit change in pressure. Intracranial compliance has limitations and decreases as intracranial pressure increases. If intracranial volume increases beyond the abilities of compensatory mechanisms, progressively larger increases in intracranial pressure result per unit of volume increase (see Figure 29-3), cerebral perfusion pressure is compromised, and ischemic death of brain tissue occurs. In cases of severe head trauma, intracranial compliance often is quickly exhausted. If mean arterial blood pressure decreases (hypotension), especially in combination with hypoxemia, the brain vasculature will vasodilate in an effort to preserve blood flow (see Figure 29-2). The increase in blood volume increases intracranial pressure, but cerebral perfusion pressure remains inadequate. In addition, the secondary autolytic processes occurring in the injured brain are enhanced by hypotension and hypoxemia, and further brain injury and edema occur with a resultant rise in intracranial pressure. • Hypertonic saline (7%): 4 to 5 mL/kg over 5 to 10 minutes for hypovolemic shock in dogs, 2 mL/kg in cats. Hypertonic saline is also available as 23.4% solution, which cannot be administered undiluted, but may be mixed 1 : 3 with hetastarch or dextran-70 (e.g., 20 mL 23.4% hypertonic saline +40 mL hetastarch or dextran-70 in a 60 mL syringe) to produce a solution of synthetic colloid suspended in a 7% hypertonic saline solution. Sodium does not freely cross the blood-brain barrier; therefore, hypertonic saline can reduce cerebral edema via an osmotic pull of fluid out of the brain parenchyma and into the intravascular space. It also has positive inotropic effects and immunomodulatory effects, and reduces endothelial swelling. Although hypertonic saline has been shown to improve mean arterial blood pressure and cerebral perfusion pressure and to protect against increased intracranial pressure, sodium has recently been implicated as the major osmotic agent contributing to brain edema. Hypertonic saline may have a global protective effect on the brain, but theoretically may lead to increased compromise to focal areas of damaged parenchyma caused by compromise of the blood-brain barrier in these regions. Because it provides rapid volume expansion and treats cerebral edema, hypertonic saline is the authors’ resuscitation fluid of choice in the euhydrated, hypovolemic head-trauma patient. Dehydrated trauma victims should receive isotonic crystalloid resuscitation. • Synthetic colloids: 10 to 20 mL/kg to effect (up to 40 mL/kg/hr) for shock. This can be given as a rapid bolus in dogs; give it in 5 mL/kg increments over 5 to 10 min in cats. Dextran-70 is an acceptable alternative but, when given as sole fluid support, has not exhibited the beneficial effects demonstrated with hetastarch and hypertonic saline. Because the volume expansion effects of synthetic colloids are limited in dehydrated patients, isotonic crystalloid fluids should be used in these cases. • Isotonic crystalloids (lactated Ringer’s solution, 0.9% saline): 20 to 30 mL/kg bolus over 15 to 20 minutes for shock. May be repeated as necessary after reassessment. Because overhydration with subsequent worsening of brain edema and increased intracranial pressure is a concern with crystalloid administration, the “shock dose” (90 mL/kg in the dog, 60 mL/kg in the cat) of crystalloids should be given incrementally to effect, as described previously. If the entire volume is not necessary to restore euvolemia and normal mean arterial blood pressure, fluid administration should be tapered when these physiologic goals are met. • Blood products: Administration of 1 mL/kg of packed red blood cells (RBCs) or 2 mL/kg of whole blood will increase the PCV by 1%. The severity of anemia will dictate the total dose to be administered, but 10 to 15 mL/kg of packed (RBCs) is a reasonable starting dose. Blood products are typically administered over 4 hours, but may be given faster (to effect) if the patient is unstable. Boluses of blood products are acceptable in the severely anemic trauma patient. The goal of therapy with blood products is to correct the anemic state to a PCV between 25% and 30%. Patients with demonstrated coagulopathy should also be treated with fresh frozen plasma at a dose of 10 to 15 mL/kg 2 to 3 times per day until coagulopathy has resolved.
Medical and Surgical Management of the Brain-Injured Pet
Brain Injury
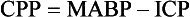
Fluid Therapy
< div class='tao-gold-member'>
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


