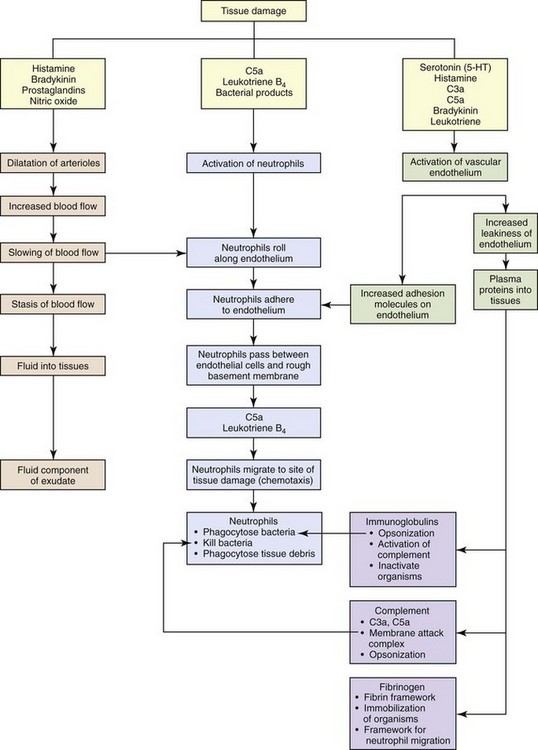
CHAPTER 3 Injury or death of cells caused by infectious microbes, mechanical trauma, heat, cold, radiation, or cancerous cells can initiate a well-organized cascade of fluidic and cellular changes within living vascularized tissue called acute inflammation (Fig. 3-1). These changes result in the accumulation of fluid, electrolytes, and plasma proteins, as well as leukocytes, in extravascular tissue and are recognized clinically by redness, heat, swelling, pain, and loss of function of the affected tissue. Inflammation is often a protective mechanism whose biologic purpose is to dilute, isolate, and eliminate the cause of injury and to repair tissue damage resulting from the injury. Without inflammation, animals would not survive their daily interactions with environmental microbes, foreign materials, and trauma and with degenerate, senescent, and neoplastic cells. WEB TABLE 3-1 Highlights of the Historical Contributions to the Understanding and Characterization of Inflammation The current understanding of inflammation has evolved over the last 3600 years of recorded history (Web Table 3-1). Clinical signs attributable to inflammation were first described in Egypt in 1600 bc. Aulus Cornelius Celsus, 25 bc to 50 ad, was a Roman writer (de Medicina) and the first individual to describe the four cardinal signs of inflammation (redness, heat, swelling, and pain) that are commonly used today to diagnose inflammation in medicine. In the mid-1800s, Rudolf Virchow, the founder of modern pathology, added the fifth cardinal sign of inflammation: loss of function. In addition to his numerous contributions to cellular pathology, Dr. Virchow, by the age of 25, had discovered fibrinogen, described the processes of leukocytosis, and later characterized pus and necrosis. In 1859, his book, Cell Pathology, became the basis for all microscopic study of disease. Phagocytosis by macrophages, an important component of inflammation and immunologic responses, was first described by Illya Mechnikov (Elie Metchnikoff) in 1883, and he was awarded the Nobel Prize in Medicine in 1908. Jules Bordet, a Belgian scientist, demonstrated antibacterial activity of serum, which Paul Ehrlich, of Germany, later defined as complement. The first experiment to demonstrate the role of a chemical mediator (histamine) in inducing vascular changes (flare and wheal reactions) was conducted by Sir Thomas Lewis in 1927. The work of these pioneers, as well as additional experimental studies conducted during the past century, has provided (1) an in-depth and clearer understanding of inflammation and (2) the foundation for development of therapeutic compounds to treat undesirable effects of inflammatory responses. In fact, such treatments are so widely used and commonplace in veterinary medicine today that the contributions and discoveries of these scientists are often taken for granted. As a general rule, inflammatory responses are beneficial in the following ways: • Diluting and/or inactivating biologic and chemical toxins • Killing or sequestering microbes, foreign material, necrotic tissue (e.g., bone sequestrum), and neoplastic cells • Providing wound healing factors to ulcerated surfaces and traumatized tissue • Restricting movement of appendages and joints to allow time for healing and repair • Increasing temperature in the body or locally to induce vasodilation and inhibiting replication of some microbial agents However, in some instances, an excessive and/or prolonged inflammatory response can be detrimental and even more harmful than that of the inciting agent/substance. In several disorders of humans, such as myocardial infarction, cerebral thrombosis and infarction, and atherosclerosis, excessive and prolonged inflammatory responses can exacerbate the severity of the disease process. In veterinary medicine, exuberant or uncontrolled inflammatory responses occurring in the diseases listed in Box 3-1 can also result in increased severity of disease. The acute inflammatory response (Fig. 3-2) can be initiated by a variety of exogenous and endogenous substances that injure vascularized tissue. The response to injury begins as active hyperemia, characterized by an increased flow of blood to injured tissue secondary to dilation of arterioles and capillaries (vasodilation), and it is this response that is responsible for redness and heat. It is facilitated by chemical mediators such as prostaglandins, endothelin, and nitric oxide (Box 3-2). With vasodilation, vascular flow is slowed (vascular congestion), allowing time for fluid leakage that occurs as a result of changes in junctional complexes of endothelial cells induced by vasoactive amines, complement components C3a and C5a, bradykinin, leukotrienes, prostaglandins, and platelet-activating factor (PAF), resulting in leakage of plasma and plasma proteins into the extracellular space (swelling and pain [stretching of pain receptors]) mainly from interendothelial cell gaps in the postcapillary venules. Fig. 3-2 The principal cellular and vascular responses during the inflammatory response. The plasma proteins and fluid that initially accumulate in the extracellular space in response to injury is classified as a transudate (Fig. 3-3). A transudate is a fluid with minimal protein (specific gravity <1.012 [<3 g of protein/dL]) and cellular elements (<1500 leukocytes/mL) and is essentially an electrolyte solution similar to that of plasma. Most commonly, the formation of a transudate occurs with hypertension, hypoproteinemia, and/or early in the acute inflammatory response. With these conditions, there is increased permeability because of small physiologic gaps between endothelial cells. Hypertension in veins and capillaries can be secondary to arterial hypertension or venous/lymphatic obstruction. Hypoproteinemia is often due to loss of albumin, the major intravascular colloidal protein, and an inability of the liver to rapidly synthesize replacement albumin. The loss of albumin allows intravascular fluid to move toward the extravascular colloids (extravascular proteins). Loss of albumin and other intracellular proteins can occur secondary to renal disease (urinary loss), severe burns, and severe hepatic disease (decreased albumin production). During the early stages of the acute inflammatory response, intercellular gaps form between endothelial cells caused by endothelial cell contraction. The gaps are very small and only allow water and electrolytes to pass through them. With persistent and widening endothelial gap formation or with endothelial cell injury, neutrophils and additional protein can enter injured areas resulting in the formation of an exudate (see Fig. 3-3). An exudate is an opaque and often viscous fluid (specific gravity >1.020) that contains more than 3 g of protein/dL and more than 1500 leukocytes/mL. As discussed in a later section, the morphologic classification of inflammatory responses into categories, such as serous, fibrinous, and/or suppurative, is based on the character of the fluid that leaks from the vessel and of the leukocytes that migrate from the vascular lumen into the extracellular space. Fig. 3-3 Formation of transudates and exudates. Fibrinogen is an important plasma protein in exudates that polymerizes in extravascular tissues to form fibrin (Fig. 3-4). Plasma dilutes the effects of the inciting stimulus, whereas polymerized fibrin confines the stimulus to an isolated area, thus preventing its movement into adjacent tissue. This confinement provides leukocytes with a well-defined target for migration during the cellular phase of the acute inflammatory response. Neutrophils are the first leukocytes to enter the exudate, and their accumulation in the exudate after they liquefy is termed pus. Neutrophils have a variety of cytoplasmic granules, such as lysosomes, that contain antimicrobial peptides and proteins, as well as matrix metalloproteinases, elastases, and myeloperoxidases. They kill pathogens and degrade foreign material by two mechanisms: (1) phagocytosis and fusion with primary and secondary lysosomes and (2) secretion of the contents of granules into the exudate. Because of the enzymes released, these cells can contribute to tissue injury. Fibrin and its products have additional activities, including chemotactic properties and blood clot formation. Fibrin also forms a framework/scaffold for fibroblast and endothelial cell migration during the initial stages of wound healing. Fig. 3-4 Examples of the appearance of fibrin in acute inflammation in the lung and mammary gland. The acute inflammatory response to either endogenous or exogenous substances occurs simultaneously with activation of the innate immune system (see Chapter 5). Innate immunity is a nonspecific defense against potentially harmful environmental substances and consists of the following: • Physical barriers and microenvironments provided by epithelia of the skin (low pH, lactic and fatty acids) and mucosae such as in the respiratory mucociliary escalator, reproductive tracts (secretions), and alimentary system (gastric and duodenal secretions, peristalsis, saliva) • Molecular products released by mucosae including lactoferrin, antimicrobial peptides (α- and β-defensins, cathelicidins), and collectins. These products have immune activity but also contribute to proinflammatory and antiinflammatory reactions, leukocyte activation, and wound healing. • Effector molecules in the blood, such as plasma proteases (complement, kinin, and clotting systems), and inflammatory mediators released from nerve fibers (sensory fibers, C-reactive fibers), such as substance P. • Effector cells in mucosae and vascularized connective tissue • Effector molecules in the blood, on endothelial cells • Visible and ultraviolet light spectra (sunburn and photosensitization) • Radiation, blunt force trauma (abrasion, bruising, incision, and laceration) • Thermal injury (hot and cold) • Microbial molecules (lipids and proteins) • Venom (insect, snake, and reptile) • Responses of the adaptive immune system (type I to IV hypersensitivities) to microbial and environmental antigens In contrast to the innate immune system, adaptive immunity results in an antigen-specific immune response characterized by the production of protective antibodies and effector leukocytes that attempt to eliminate the inciting cause of injury and the generation of memory cells that make subsequent adaptive immune responses against a specific microbial antigen more efficient and effective (see Chapter 5). Because acute inflammation is a vasocentric response, it would be reasonable to assume that just about any exogenous or endogenous cause could induce inflammation. This is true, but because the inflammatory response has numerous redundant checks and balances that regulate the occurrence and severity of expression of the response, harmful effects of inflammation are minimized. • Vasoactive amines such as histamine and serotonin • Plasma proteases such as complement, kinin, and clotting system proteins • Lipid mediators such as arachidonic acid metabolites and platelet-activating factor (PAF) • Increased blood flow (active hyperemia) to the site of injury • Increased permeability of capillaries and postcapillary venules to plasma proteins and leukocytes through release of inflammatory mediators • Emigration of leukocytes (via the leukocyte adhesion cascade) into the perivascular area Initially, arterioles dilate and capillary beds in the affected area expand in volume to accommodate an increased blood flow (heat and redness) in response to the stimulus. Second, as a result of permeability changes induced by inflammatory mediators, blood flow through the capillary beds is slowed as a result of increased viscosity and hemoconcentration after leakage of water from capillary beds into extracellular space. Because of the reduced blood flow and pressure, microscopically, capillaries are often packed with erythrocytes and the microenvironment facilitates leukocytic margination along the luminal surface of endothelial cells. This stage precedes leukocyte emigration through intercellular junctions of endothelial cells into the extravascular space. Inflammatory mediators induce endothelial cell contraction, resulting in the formation of interendothelial cell gaps, which further allow fluid leakage and leukocyte migration. In general, the endothelium of the normal vascular capillaries limits exchange of molecules to those less than 69,000 MW, the size of albumin. The exchange of small molecules and water between the vessel lumen and the interstitial space is extremely rapid. For example, the water of plasma is exchanged with the water of the interstitial space 80 times before the plasma can move the entire length of the capillary. Physiologically, increased amounts of fluid can pass across the vascular wall when there is (1) excessive hydrostatic pressure caused by hypertension and/or sodium retention, (2) decreased plasma proteins (colloid), or (3) lymphatic and/or venous obstruction. If fluid leakage is excessive, edema (transudate) develops (see Fig. 3-3). If the leakage is not excessive and postcapillary venules and lymphatic vessels are functioning normally, all of the fluid released from arterioles and small capillaries is returned to the circulation via paracellular gaps of postcapillary venular and lymphatic vessels. During acute inflammatory responses, there is a net outflow of fluid from arterioles, capillaries, and venules into extracellular tissue, which overwhelms the capacity for resorption by postcapillary venules and lymphatic vessels). The mechanisms of leakage (Fig. 3-5) depend on the biologic and physical characteristics of the inciting agent or substance and include the following: Fig. 3-5 Principal mechanisms of increased vascular permeability in inflammation and their features and underlying causes. • Opening of junctional complexes (endothelial gaps) between endothelial cells responding to inflammatory mediators • Direct injury that results in necrosis and detachment of endothelial cells, as occurs with certain viral, protozoal infections, toxins, and radiation • Leukocyte-dependent injury that results in necrosis and detachment of endothelial cells and is induced by enzymes and mediators released from leukocytes during the transmigration phase of the acute inflammatory response • Increased endothelial cell transcytosis mediated by vascular endothelial growth factor (VEGF) The movement of leukocytes from the lumina of capillaries and postcapillary venules into the interstitial connective tissue occurs through a process called the leukocyte adhesion cascade (Fig. 3-6). Chemokines, cytokines, and other inflammatory mediators influence this process by modulating the surface expression and/or avidity of adhesion molecules on both endothelial cells and leukocytes. It has a well-characterized sequence of events, including margination, rolling, activation and stable adherence (adhesion), and transmigration of leukocytes toward a chemotactic stimulus. Fig. 3-6 Leukocyte adhesion cascade. • Margination. As vessels vasodilate and have reduced hydrostatic pressure and blood flow, leukocytes exit the central region of the vascular lumen and move to the periphery of the vascular lumen near the endothelial cell surface (marginization). • Rolling. The initial contact between leukocytes and endothelial cells occurs by transient, weak binding interactions between the selectin family of adhesion molecules and their receptors. During rolling, leukocytes temporarily bind to endothelium and then release, which brings the leukocyte close to endothelial cell surface and reduced velocity of the traveling leukocyte. This process is mediated by selectins, including L-selectin expressed by neutrophils and P-selectin, a carbohydrate binding molecule stored in Weibel-Palade bodies of endothelial cells and α-granules of platelets, as well as E-selectin expressed by endothelial cells. L-selectin is expressed on all leukocytes, but at low concentrations in normal human neutrophils and binds sialyl Lewis X receptor (and other receptors) on endothelial cells. P-selectin molecules expressed on endothelial cell surfaces bind to P selectin glycoprotein ligand 1 (sialyl Lewis X–modified proteins) present on neutrophils, eosinophils, monocytes, and lymphocytes. E-selectin also mediates leukocyte-endothelial cell adherence and is expressed on endothelial cell surfaces for binding glycoprotein receptors expressed on leukocytes. Selectin-mediated attachments are formed at the leading edge of the rolling leukocyte and broken at the trailing edge. Even slight disturbances, such as surgical manipulation, heat, temporary ischemia, and mast cell products, induce rolling of neutrophils along the surface of endothelial cells. By slowing leukocyte transit time through capillaries and postcapillary venules, combined with the continual close proximity of slow rolling leukocytes to the endothelium, and the continued release of chemokines and proinflammatory cytokines, the proper microenvironment for progression to the “stable adhesion” stage occurs. • Stable adhesion. For stable adhesion to occur, neutrophils and endothelial cells become activated by a variety of cytokines (such as IL-1, interleukin-6 [IL-6], TNF), complement factors (C5a), PAF, platelet-derived growth factor (PDGF), chemokines, and other inflammatory mediators. Once neutrophils are activated, L-selectin molecules are proteolytically cleaved from the neutrophil surface by ADAM17, and the neutrophils express a new set of membrane proteins (integrins) by rapid exocytosis of cytoplasmic vesicles. Firm adhesion of neutrophils to endothelial cells is mediated by binding of β2 integrin molecules, such as Mac-1 (CD11a/CD18), that are expressed on stimulated neutrophils in an active conformation, to intercellular adhesion molecule-1 (ICAM-1) and other ICAM molecules on endothelial cells. P- and E-selectin adherence also contributes to the process of firm adhesion. There are four β2 integrins (lymphocyte function antigen-1 [LFA-1], Mac-1, p150,95, and αdβ2), each of which are heterodimers that differ only in their subunit CD11 a, b, c, and d for LFA-1, Mac-1, p150,95, and αdβ2, respectively. CD18 (the β-subunit) is identical in all four β2 integrins. Three β2 integrins (LFA-1, Mac-1, and p150,95) are involved with leukocyte adherence; however, the β2 integrin αdβ2, which was first identified in dogs and subsequently in humans, is apparently not meaningfully involved with the adherence of neutrophils or other leukocytes to endothelium. Once stable adherence is achieved, neutrophils move to endothelial cell junctions for transendothelial cell migration. • Transendothelial cell migration. In postcapillary venules, neutrophil movement decreases from 10 µm/sec to a complete stop after margination. Firmly adhered leukocytes emigrate (transmigrate) across the endothelial layer by passing between endothelial cells. A number of leukocyte adhesion molecules are involved in this process (Table 3-1). Adhesion molecule activity and expression differ slightly for different tissues and cell types. In noninflamed skin, for example, there is a higher level of E- and P-selectin expression on endothelial cells, which facilitates rolling of leukocytes. In inflamed liver, CD44 of neutrophils binds serum-derived hyaluronan-associated protein (SHAP) that is bound to hyaluronic acid present on the luminal surface of endothelial cells. Nonactivated lymphocytes and monocytes utilize L-selectin to mediate adherence to high endothelial venules (HEVs) in lymph nodes. These cells also utilize α4β1 (very-late antigen-4 [VLA-4]) to mediate stable adherence to the endothelial ligand, vascular cell-adhesion molecule-1 (VCAM-1). Neutrophils and other leukocytes transmigrate between endothelial cells at the intercellular junctions. Platelet-endothelial cell adhesion molecule-1 (PECAM-1), a molecule that is present on endothelial cell membranes, and junctional adhesion molecules (JAM) A, B, and C mediate adherence activities and the adherence process. Contributions to this process also include β2 integrin binding of ICAM-1 and E-selectin binding. Pseudopodia of neutrophils and other leukocytes extend between endothelial cells and come into contact with and bind to the basement membrane (composed of laminin and collagens) and subjacent extracellular matrix (ECM) proteins (proteoglycans, fibronectin, and vitronectin). This binding interaction is mediated, at least in part, by the β1 integrins. Neutrophils that pass across the vascular wall accumulate in the perivascular connective tissue stroma within the inflammatory exudate. Once within the perivascular stroma, neutrophils migrate along a pathway established by the chemotactic gradients and inflammatory mediators. TABLE 3-1 Endothelial Cell/Neutrophil Adhesion Molecules From Cotran RS, Kumar V, Collins T, et al: Robbins pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders. This process is actually initiated during the fluidic phase of the acute inflammatory response and is driven by chemokines, cytokines, and chemoattractant substances such as complement. Temporally, margination, rolling, activation and firm adhesion, and transmigration all occur concurrently, involving different leukocytes in the same capillaries and postcapillary venules. This process is largely mediated by the interaction of ligands expressed on the surface of neutrophils, lymphocytes, and macrophages and their receptors expressed on luminal surfaces of activated endothelial cells (see Table 3-1). Adhesion molecules are divided into (1) selectins (E-, L-, and P-selectin), (2) integrins (VLA family of β1 integrins; β2 integrins [Mac-1, LFA-1, p150,95, αdβ2]), (3) cytoadhesin family (vitronectin, β3 integrins, and β7 integrins used predominately by lymphocytes), (4) the immunoglobulin superfamily (ICAM-1 to ICAM-3, VCAM-1, PECAM-1) and mucosal addressin adhesion molecule-1 (MAdCAM-1), and (5) other molecules such as CD44 (Web Table 3-2). WEB TABLE 3-2 Summary of Various Leukocyte Adhesion Molecules This table is not comprehensive for cellular expression and binding activity. WEB TABLE 3-3 Leukocyte Defects Identified in Animals and Humans NADPH, Nicotinamide adenine dinucleotide phosphate. From Cotran RS, Kumar V, Collins T, et al: Robbins pathologic basis of disease, ed 7, Philadelphia, 2005, Saunders. Web Fig. 3-1 Bovine leukocyte adhesion deficiency (BLAD), gross and microscopic characteristics. Web Fig. 3-2 Reduced neutrophil infiltration in lung of a calf with bovine leukocyte adhesion deficiency (BLAD). Web Fig. 3-3 Bovine leukocyte adhesion deficiency (BLAD), intravascular neutrophils partially adhered (rolling) on endothelial cells, capillary, alveolar septum, lung. The three types (I, II, and III) of leukocyte adhesion deficiencies (LADs) are the result of one or more defects in the sequence of steps leading to migration of leukocytes into the site of inflammation. These deficiencies are rare in humans, cattle, and dogs (Web Table 3-3), although debilitation often occurs in humans and animals with severe forms of LADs. These clinical conditions underscore the importance of leukocytes in host defense and the vital role that these adhesion molecules serve in the transmigration process. Cattle, dogs (Irish setters), and humans with LAD type I lack functional expression of the β2 integrins. Affected individuals often have high numbers of neutrophils, exceeding 125,000/µL in the blood; however, in cattle, these neutrophils have impaired passage across vascular walls (Web Fig. 3-1) and numerous other functional abnormalities related to inadequate membrane adherence activity. As a result, lesion sites lack significant neutrophils. Cattle with bovine LAD (BLAD) develop severe oral ulcers, gingivitis, tooth loss, enteric ulcers, cutaneous ulcers, abscesses that lack pus formation, and pneumonia (see Web Fig. 3-1). Most affected cattle die within few days or weeks after birth as the result of diarrhea and/or pneumonia. The hallmark lesion histologically is a sparse infiltration of neutrophils into ulcerated mucosal surfaces or pulmonary alveoli, despite high numbers of neutrophils within lumen of submucosal and pulmonary septal blood vessels (Web Figs. 3-2 and 3-3). There are no effective treatments for calves and Irish setters with LAD, although cattle with BLAD can survive into adulthood with intensive medical care. The deficiency in β2-integrin expression in Holstein calves is due to a single-point mutation (adenine → guanine) at position 128 resulting in a single amino acid change (aspartic acid → glycine) in the β-subunit (CD18) of the β2 integrins. This defect occurs in a highly conserved extracellular region (amino acids 96 through 389) of the protein. A second, silent mutation has also been detected in cattle with BLAD and does not alter the amino acid sequence. In Irish setters with canine LAD (CLAD), there is a single missense mutation resulting in a G to C transversion at nucleotide 107 of the cDNA sequence, resulting in a serine that replaces a highly conserved cysteine. In 1992, a second form of LAD was discovered in humans and called LAD type II (see Table 3-3). In LAD type II, patients lack fucosylated sialyl Lewis X used for selectin-mediated adherence caused by a mutation in a fucose transporter gene. Their CD18 binding remains intact; however, these patients suffer from the same types of lesions and infections, as do patients with CD18 deficiency. Therefore these observations demonstrate the importance of CD18-independent adherence. More recently, a third type of LAD (LAD type III) has been identified. LAD type III is caused by a defect in kindlin-3, which is a cytoplasmic protein essential for integrin activity in the cytosol. LAD type III patients have defects in leukocyte function and platelet aggregation. Both LAD types II and III have not been identified in animals. The role of mast cells and basophils in IgE-mediated hypersensitivity reactions has been known for decades. These cells are critical effector cells in disorders of IgE-dependent immediate type I hypersensitivities (see Chapter 5). The release of their granules and mediators at inflammatory concentrations in the lung, for example, results in mucus secretion, accumulation of seroproteinaceous fluid in airways, bronchoconstriction, and vasodilation. The excessive release of tryptase and chymase by mast cells may enhance degradation of the ECM, which contributes to fibrosis and tissue remodeling. Chemokines and cytokines from mast cells and basophils contribute to innate immune defenses through chemotaxis and release of antimicrobial peptides. The mediators also enhance adhesion molecule expression on endothelial cells of nearby blood vessels and leukocytes that enter the area.
Inflammation and Healing
Contributor
Time
Contribution
Egyptians
1600 bc
Descriptions of inflammation
Celsus (Italy)
25 bc-50 ad
Four cardinal signs of inflammation: rubor (redness), tumor (swelling), calor (heat), dolor (pain)
John Hunter (Scotland)
1793
Inflammation is a salutary (favorable to health) effect, not a disease per se
Julius Cohnheim (Germany)
1839-1884
Observed inflamed vessels microscopically
Elie Metchnikoff (Russia)
1882
Observed and described phagocytosis
Rudolf Virchow (Germany)
1821-1902
Fifth cardinal sign of inflammation: functio laesa (loss of function); cellular injury
Sir Thomas Lewis (England)
1927
Determined that chemicals (histamine) induce vascular changes
Jules Bordet (Belgium)
1898
Antibacterial effects of serum
Paul Ehrlich (Germany)
1908
Complement mediates antibacterial effects of serum
Evolution of the Current Understanding of Inflammation
Beneficial and Harmful Aspects of Inflammation
Acute Inflammation
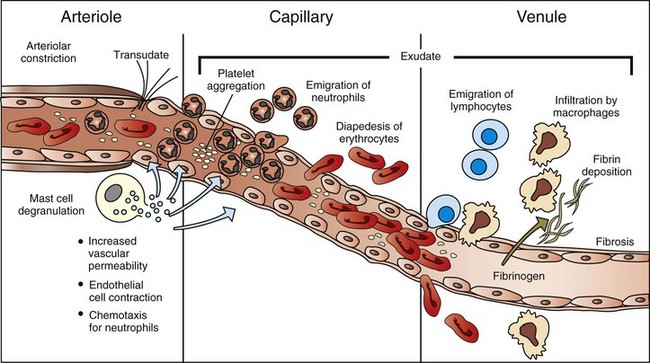
The majority of leukocyte transmigration and hemorrhage occurs in the capillaries and postcapillary venules. (Modified from McCance KL, Huether SE: Pathophysiology: the biologic basis for diseases in adults and children, ed 3, St Louis, 1998, Mosby.)
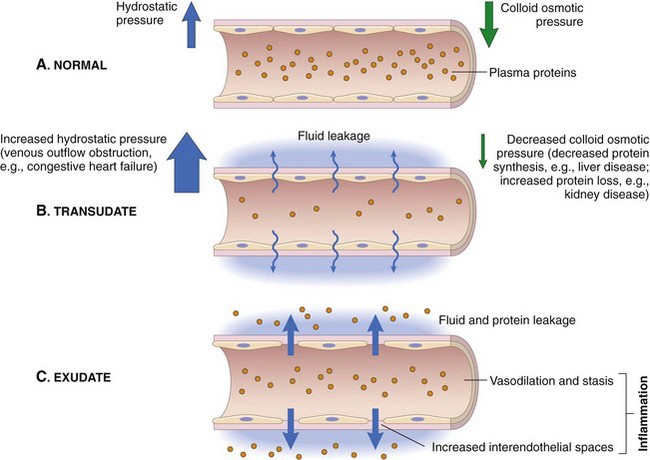
A, Normal hydrostatic pressure (blue arrows) is about 32 mm Hg at the arterial end of a capillary bed and 12 mm Hg at the venous end; the mean colloid osmotic pressure of tissues is approximately 25 mm Hg (green arrows), which is equal to the mean capillary pressure. Therefore the net flow of fluid across the vascular bed is minimal. B, A transudate forms when fluid leaks out because of increased hydrostatic pressure or decreased osmotic pressure. C, An exudate forms in inflammation because vascular permeability increases as a result of increased interendothelial spaces. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
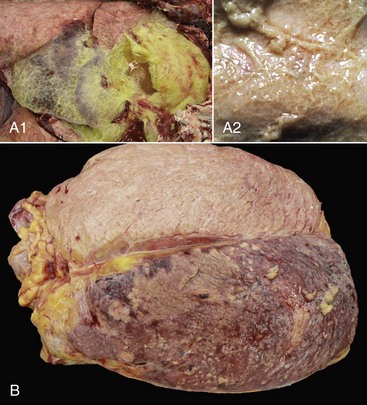
A1, Lungs (in situ) from an ox with acute pleuritis. The lungs have a thick mat of fibrin covering the cranioventral region. A small area of fibrin is torn away revealing the subjacent clear, yellow fluid subjacent. The fibrin coating the lung surface was released as fibrinogen from inflamed pleural vessels and polymerized on the serosal surface. The remaining lung surface is less affected. A2, Higher magnification of fibrin on the surface of the lung. B, Acute fibrinonecrotic mastitis, mammary gland, horizontal section, cow. The left quarters of the mammary gland (lower half of the image) are swollen as a result of edema and fibrin exudation and are reddened because of hyperemia, vascular congestion, and hemorrhage. The right quarters (upper half of the image) are unaffected. (A1 and B courtesy Dr. J.S. Haynes, College of Veterinary Medicine, Iowa State University; A2 courtesy Dr. M.D. McGavin, College of Veterinary Medicine, University of Tennessee.)
Substances Inducing the Acute Inflammatory Response
Fluidic (Exudative) Phase of the Acute Inflammatory Response
Endothelial Cell Dynamics During the Acute Inflammatory Response
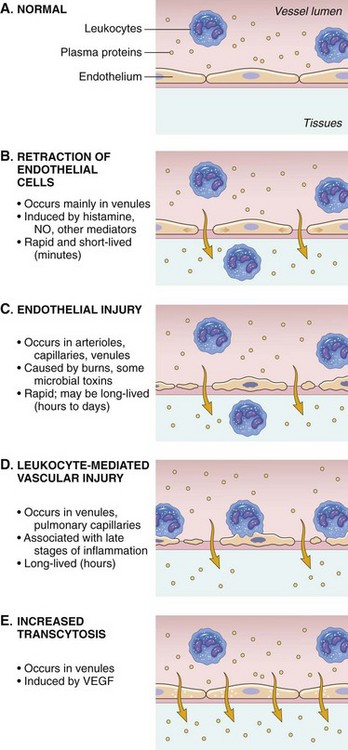
NO, Nitric oxide; VEGF, vascular endothelial growth factor. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Cellular Phase of the Acute Inflammatory Response
Leukocyte Adhesion Cascade
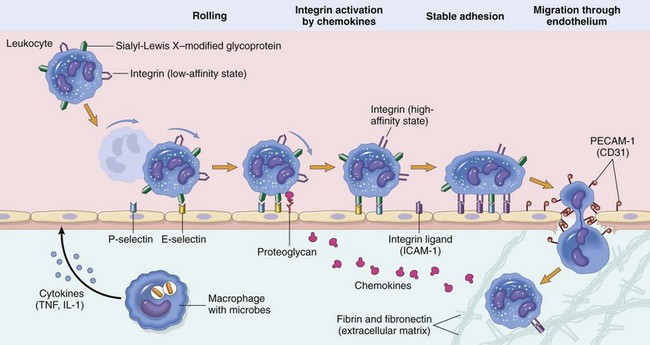
Steps involved with neutrophil migration across the vascular wall include rolling, activation, stable adhesion, and migration across the endothelial layer and vascular wall. Neutrophils express receptors for E- and P-selectin, which bind to ligands expressed on endothelial cells. L-selectin is also expressed by bovine neutrophils and to a varying degree in other species. The rolling process slows neutrophil movement within the vessel and brings the neutrophil closer to the vascular endothelial cell surface. Activation is induced by inflammatory mediators, including chemokines, such as interleukin-8 (IL-8), and cytokines, such as interleukin-1β (IL-1β) and tumor necrosis factor-α (TNF-α), which are released by nearby leukocytes and endothelial cells. These activate both neutrophils and endothelial cells which then changes the conformation of integrins allowing for enhanced binding avidity to integrin receptors and increased expression of additional adhesion molecules. Binding of β2 integrins (Mac-1; CD11a/CD18) expressed by leukocytes to intercellular adhesion molecule-1 (ICAM-1) expressed by endothelial cells leads to stable adherence of the cell to the endothelial cell surface. Once leukocytes are attached to the vascular endothelium, they adhere to platelet-endothelial cell adhesion (PECAM-1) and other junctional adhesion molecules present at the endothelial cell junction and transmigrate through the junction into the perivascular tissue, where they express β1 integrins that adhere to extracellular matrix proteins such as laminin, fibronectin, vitronectin, and collagen. The process is mediated by chemokines (CXCL8; IL-8), complement fragments, vasoactive amines, cytokines, and membrane-derived mediators such as platelet-activating factor (PAF) and leukotrienes. (From Kumar V, Abbas A, Fausto N, et al: Robbins & Cotran pathologic basis of disease, ed 8, Philadelphia, 2009, Saunders.)
Endothelial Molecule
Leukocyte Receptor
Major Role
P-selectin
Sialyl-Lewis X
PSGL-1
Rolling (neutrophils, monocytes, lymphocytes)
E-selectin
Sialyl-Lewis X
ESL-1, PSGL-1
Rolling, adhesion to activated endothelium (neutrophils, monocytes, T-lymphocytes)
ICAM-1
CD11/CD18 (integrins)
(LFA-1, Mac-1)
Adhesion, arrest, transmigration (all leukocytes)
PECAM-1
PECAM-1
Transendothelial cell migration
JAM A
JAM A, LFA-1
Transendothelial cell migration
JAM C
JAM B, Mac-1
Transendothelial cell migration
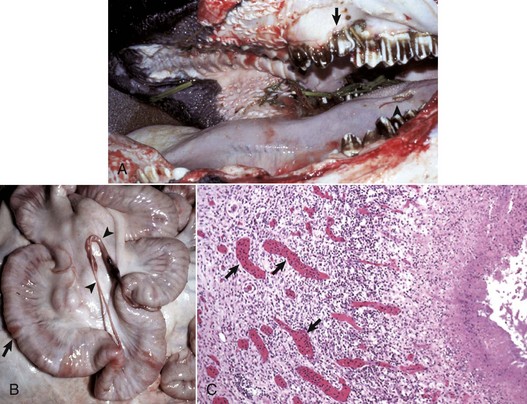
A, The oral cavity has irregularly arranged molar teeth (arrow), oral ulcers (arrowhead), and grass material within the oral cavity secondary to impaired mastication. B, The serosa of the small intestine is thickened (arrow), and there are fibrous tags between serosal surfaces (arrowheads). C, Microscopically the mucosa underlying the areas of thickened serosa is ulcerated and covered by cell debris. Vascular lumina contain elevated numbers of neutrophils, which do not adhere to the vascular endothelium (arrows) despite the ulcerated and “inflamed” mucosa. Lymphocytes, macrophages, and plasma cells are present in perivascular areas of Peyer’s patches, but the surrounding lymphoid tissue virtually lacks neutrophils. The lack of neutrophil adherence to the vascular endothelium and of infiltration into the perivascular areas is caused by the lack of expression of β2 integrins in BLAD. The lumen of the intestine is to the right. H&E stain. (Courtesy Dr. M.R. Ackermann, College of Veterinary Medicine, Iowa State University.)
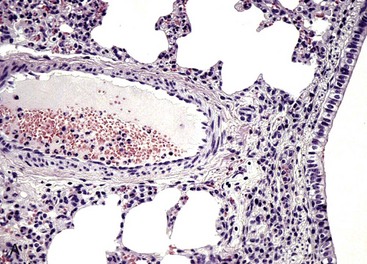
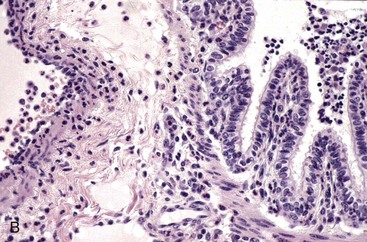
Both lungs were inoculated with Mannheimia haemolytica and in the animal with normal CD18 expression, neutrophils pass across the vascular wall into the extravascular space and the nearby airway lumen. In contrast, neutrophils in the calf with BLAD are confined to the vascular lumen and do not pass across the vascular wall. Thus the lack of CD18 expression impaired neutrophil stable adherence and migration across the vascular wall, despite the bacterial stimulus. (Courtesy Dr. M.R. Ackermann, College of Veterinary Medicine, Iowa State University.)
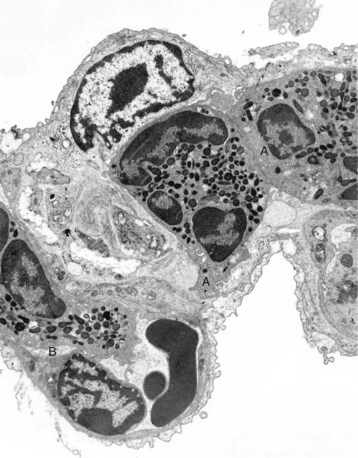
The septum is thickened because of two neutrophils (A) within the septal wall and a neutrophil (B) within the lumen of a vascular capillary (adherence). Transmission electron microscopy (TEM). Uranyl acetate and lead citrate stain. (Courtesy Dr. M.R. Ackermann, College of Veterinary Medicine, Iowa State University.)
Leukocyte Adhesion Deficiencies
Effector Cells of the Acute Inflammatory Response
Mast Cells and Basophils
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Veterian Key
Fastest Veterinary Medicine Insight Engine