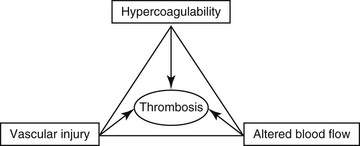
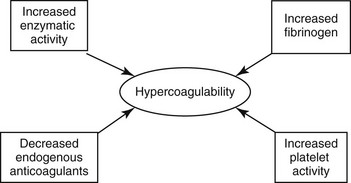
Chapter 64 A hypercoagulable state is an enhanced tendency to form venous or arterial thrombi. In 1845 a young German pathologist named Rudolf Virchow presented a novel and relatively simple concept of the pathogenesis of thromboembolic disease, which remains valid to this day. Virchow described three factors (Figure 64-1) that are the core reasons for the development of venous thrombosis: alterations of the blood (hypercoagulability), impairment of blood flow (stasis), and changes in the vessel wall (vascular injury). Research into the molecular basis of thrombosis has since enhanced our understanding of the pathophysiology of each of these concepts and thereby also our ability to diagnose and treat patients with thromboembolic disease. This chapter focuses primarily on the first of the three legs of Virchow’s triad—hypercoagulable states—but briefly describes the effects of stasis and disruption of the intact endothelium. Figure 64-1 Virchow’s triad, which describes the three factors contributing to the development of venous thrombosis. Primary (hereditary) deficiencies, such as factor V Leiden, prothrombin G20210A mutation, deficiencies of natural anticoagulants (antithrombin, protein C, and protein S), and hyperhomocysteinemia, have not been described in animals, but there are a number of acquired underlying conditions or disease states that are associated with increased risk of thrombosis in dogs and cats. The causes of secondary hypercoagulable states often are unclear and may be multifactorial but can be divided into the following categories (Figure 64-2): (1) decreased levels of endogenous anticoagulants, (2) increased enzymatic activity, (3) increased fibrinogen levels, and (4) increased platelet activity.
Hypercoagulable States
Causes
< div class='tao-gold-member'>
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


