Chapter 103 Hepatic Encephalopathy
CAUSES
In dogs and cats, congenital extrahepatic or intrahepatic portal-to-systemic venous communications are the most frequent cause of HE; up to 95% of affected animals demonstrate neurologic symptoms.1 These communications are generally a single vessel, but multiple extrahepatic2 and intrahepatic3 congenital portosystemic shunts have been reported. Hepatic arteriovenous malformations cause portal hypertension, multiple extrahepatic portosystemic shunts, and ascites, and may cause symptoms of HE. In young dogs, hepatic microvascular dysplasia4 and, rarely, congenital urea cycle deficiencies5 can also cause symptoms of HE. In older animals, portosystemic shunts develop secondary to portal hypertension that results from chronic liver disease. In cats, hepatic lipidosis often is associated with symptoms of HE. Other causes of chronic and acute hepatic failure that can result in symptoms of HE are discussed in Chapter 127, Hepatic Failure.
PATHOPHYSIOLOGY
In 1893, Marcel Nencki and Ivan Pavlov described the physiologic consequences of a surgically created, end-to-side portocaval shunt (Eck fistula) and showed that clinical signs in this canine model worsened after a meat meal, linking HE to the concept of “meat intoxication.”6 Ever since this description, HE has been thought of as a condition caused by gut-derived toxins that are not metabolized by a diseased or failing liver. Research over the last century has elaborated on this concept and demonstrated the complexity of this condition. However, recent work on several aspects of HE including cerebrospinal fluid amino acid alterations,7,8 glutamate neurotoxicity,9 the generation of reactive oxygen species,10 and the mitochondrial permeability transition11 emphasizes the central role of elevated blood ammonia concentrations in animals with HE. Other substances that are considered synergistic with ammonia toxicity include mercaptans, free fatty acids, phenols, and bile salts12 (see Chapter 127, Hepatic Failure, Table 127-2 for a summary of toxins implicated in HE).
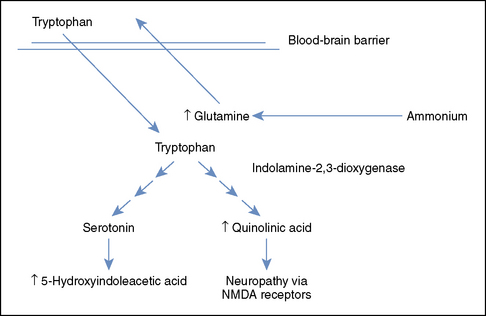
The brain has no urea cycle; consequently, ammonia in the central nervous system (CNS) is removed by transamination of glutamate into glutamine in astrocytes.16 Glutamine concentrations in the cerebrospinal fluid are elevated in dogs with HE8 and often are an accurate indicator of the degree of neurologic dysfunction in humans with HE.9 Glutamine is exchanged across the blood-brain barrier for tryptophan, leading to increased levels of tryptophan and tryptophan metabolites in the CNS (Figure 103-1).7 The tryptophan metabolites serotonin and quinolinate are important agonists of inhibitory and excitatory neurotransmission, respectively, although the exact alterations in both of these systems in patients with HE are complex and incompletely understood. Glutamine is also transported from astrocytes into neurons, where it is converted to glutamate.17 Overstimulation of the N-methyl-D-aspartate receptors by both glutamate and ammonia can cause seizures and neurotoxicity, in part as a result of free radical formation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


