Fig. 1.
Histopathology of a representative human pilocytic astrocytoma (WHO grade I). (a) Glial fibrillary acidic protein (GFAP) immunostaining demonstrates the presence of neoplastic glial cells. (b) Hematoxylin and eosin (H&E) stain reveals brightly staining Rosenthal fibers (arrow) and dark eosinophilic granular bodies (arrowhead).
2.2 Molecular Genetics
There are few genetic alterations observed in sporadic PAs. Oncogenic KRAS (19, 20) and TP53 (21–23) mutations have been identified, though these are rare events. Array-based comparative genomic hybridization studies have revealed copy number gains on chromosome 7q34, which includes the region spanning the BRAF locus. Further studies established that this gain represents a tandem duplication, resulting in fusions between an unknown gene (KIAA1549) and BRAF (1–3). The fusion transcripts contain the amino terminus of the KIAA1549 gene and the BRAF kinase domain, creating a new molecule in which unregulated BRAF function leads to increased mitogen-activated protein kinase (MAPK) signaling.
Various studies have now confirmed that BRAF rearrangement is specific for sporadic PA, reinforced by the observation that two-thirds of sporadic PAs contain this fusion protein. This is in striking contrast to patients with NF1 who lack this rearrangement and instead harbor inactivation of the NF1 tumor suppressor gene (1, 24). While this signature mutation represents a common genetic event in sporadic PA, the exact role of BRAF fusion protein expression in gliomagenesis is currently unknown. In this regard, the KIAA:BRAF fusion protein exhibits constitutive RAF kinase activity and can transform NIH3T3 fibroblasts; however, BRAF activation alone is not sufficient to drive glioma formation in mice (25). The fact that BRAF activation alone is not tumorigenic suggests that additional conditions must be required for glioma formation.
3 NF1-Associated Pilocytic Astrocytomas
3.1 Clinical Features
NF1 is an autosomal dominant condition occurring in one in 2,500–3,000 individuals worldwide (26). Children with NF1 often present with pigmentary features (café-au-lait spots, skinfold freckling, iris hamartomas), cardiovascular problems, orthopedic abnormalities (scoliosis, pseudarthrosis of the long bones), and learning disabilities. Importantly, individuals with NF1 have a predisposition to the development of peripheral and central nervous system tumors. Cutaneous and subcutaneous neurofibromas are benign peripheral nerve sheath tumors that develop in nearly all adults with NF1. One-third of individuals with NF1 will form a diffuse plexiform neurofibroma. These benign, but often extensive tumors, can impinge on neighboring nerves, blood vessels, or structures, and occasionally transform to malignant tumors (27). Upon transformation, their malignant counterparts, termed malignant peripheral nerve sheath tumors (MPNST), frequently result in metastasis and death (28).
Low-grade gliomas are the most common central nervous system tumor seen in children with NF1, and in contrast to their sporadic counterparts, most often develop along the optic pathway, including the optic nerves, chiasm, and hypothalamus (29, 30) (Fig. 2). Less commonly, gliomas may also arise in the brainstem or cerebellum (31). Optic pathway gliomas (OPG) can be asymptomatic (32, 33), but can result in decreased visual acuity, proptosis, and endocrine abnormalities (34–37). Compared to sporadic PAs, NF1-associated PAs are typically discovered earlier in childhood, with the majority being diagnosed within the first 6 years of life (32, 38). These NF1-associated optic gliomas usually have a more indolent clinical course and are less likely to progress or require treatment (39). There are rare reported cases of spontaneously regression (40, 41).
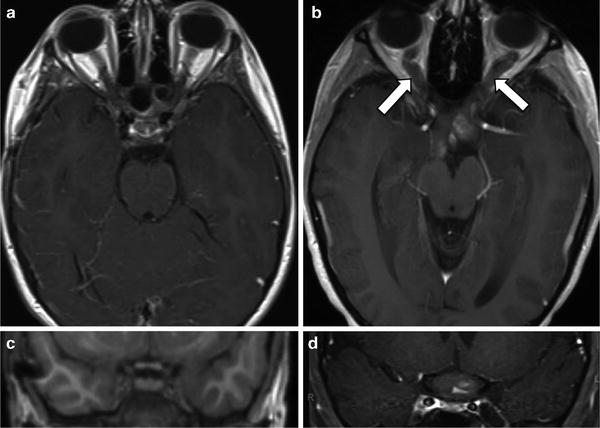
Fig. 2.
MRI of a representative NF1-associated optic pathway glioma. Magnetic resonance images of the (a) optic nerves and (c) optic chiasm (magnified) from a child without an optic glioma are shown. By contrast, MRI reveals (b) an optic pathway glioma with expansion of the optic nerves (arrows) and (d) optic chiasm.
Treatment of NF1-associated optic gliomas rarely involves surgery, which is typically reserved for cosmetic purposes in a blind eye or for debulking large tumors that exert mass effect or cause hydrocephalus. Radiation therapy is avoided in individuals with NF1 due to the six- to eightfold increased risk of developing radiation-induced secondary malignant brain tumors as well as an elevated incidence of post-radiation neuropsychological problems and cerebral vasculopathies (moya-moya syndrome) (42, 43). Chemotherapy with carboplatin and vincristine is the first-line treatment for progressive symptomatic NF1-associated OPGs (44–46), and other agents, including vinblastine and temozolomide, represent second-line therapies for progressive tumors.
3.2 Neurofibromin Growth Control
The NF1 gene on chromosome 17 encodes a large, 220–250 kDa, cytoplasmic protein, termed neurofibromin, which is expressed in many different cell types in the brain. Inspection of the predicted protein sequence of the NF1 gene revealed that a small portion of the protein contained a 300 amino acid domain which functions as a GTPase activating protein (GAP) molecule (47), thereby establishing neurofibromin as a negative regulator of the Ras proto-oncogene. Similar to other GAP molecules, neurofibromin promotes the conversion of active GTP-bound Ras to inactive GDP-bound Ras, which reduces activation of downstream signaling molecules and attenuates cell growth (Fig. 3). In this regard, elevated Ras activity is mitogenic for many cell types, prompting early NF1-associated plexiform neurofibroma clinical trials using inhibitors of farnesyltransferase (FTPIs) to block Ras activity.
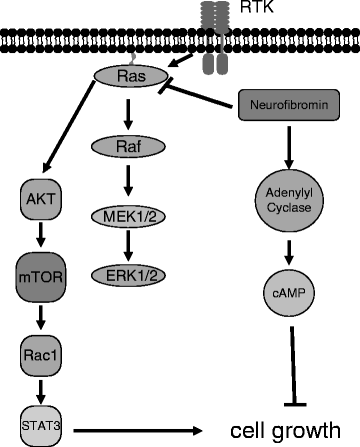
Fig. 3.
Neurofibromin-regulated intracellular signaling pathways. The NF1 gene product, neurofibromin, accelerates the conversion of the Ras proto-oncogene from its active GTP-bound form to its inactive GDP-bound form. GTP-bound Ras activates downstream signaling intermediates, including Raf/MEK/ERK and AKT/mTOR, leading to increased cell growth. Neurofibromin also increases cAMP activity through adenylyl cyclase to inhibit astrocyte cell growth.
Further insights into the mechanism by which neurofibromin regulates cell growth in the brain have derived from studies using Nf1 GEM models. Studies from our laboratory established mammalian target of rapamycin (mTOR) as a key Ras downstream signaling molecule critical for controlling Nf1-deficient astrocyte cell growth (48, 49). These studies demonstrated that the mTOR pathway may be a target for biologically based NF1-associated brain tumor therapies. Using Nf1 optic glioma GEM strains, rapamycin, a macrolide antibiotic that inhibits mTOR activity, reduces tumor growth in vivo (50). Based on these promising results, clinical trials using rapamycin analogs are currently underway for patients with NF1-associated optic glioma.
Using high-throughput screening technologies, another Ras target downstream was identified. Nf1-deficient astrocytes have increased signal transducer and activator of transcription 3 (STAT3) activation, resulting from high levels of mTOR signaling (51). STAT3 regulates the transcription of numerous genes essential for cell cycle progression, including cyclin D1 (51, 52). While nonspecific STAT3 inhibitors have demonstrated a reduction in Nf1-deficient astrocytes in vitro and attenuated NF1-deficient glioma growth in vivo, more selective STAT3 inhibitors will need to be developed for use in children with NF1-associated OPG.
An additional mechanism by which neurofibromin deficiency leads to tumor development is through defective generation of intracellular cyclic-adenosine monophosphate (cAMP) (Fig. 3). Neurofibromin positively regulates cAMP production at the level of adenylyl cyclase (AC) (53), such that in the absence of neurofibromin, the lower intracellular cAMP levels enhance cell survival in response to chemokines (54). This enhanced survival of Nf1−/− astrocytes to chemokines, like CXCL12, is the opposite of what occurs in wild-type astrocytes, where CXCL12 treatment leads to increased apoptosis (54). The attenuated cAMP levels in Nf1-deficient astrocytes provide another therapeutic opportunity. Inhibition of cAMP degradation using Rolipram, a phosphodiesterase 4 (PDE4) inhibitor, restores cAMP levels in Nf1-deficient astrocytes in vitro and attenuates optic glioma growth in vivo (55, 56). Based on these encouraging findings, future strategies for treating NF1-associated gliomas may involve a combination of cAMP- and Ras/AKT/mTOR-targeted therapies.
3.3 Nf1 GEM Models of Optic Glioma
Initial attempts at mouse modeling entailed the creation of mice lacking Nf1 gene expression in all cells in their body. These Nf1−/− mice died in utero (57, 58), precluding their use for tumor modeling. However, Nf1 conditional knockout mice developed by Dr. Luis Parada and his colleagues have enabled researchers to generate mice harboring Nf1 inactivation in specific cell populations. Using this Nf1 conditional knockout approach, previous studies revealed that loss of neurofibromin expression in central nervous system astrocytes results in increased cell proliferation, but is not sufficient for glioma formation (59). Since children with NF1 are born with one nonfunctional and one functional copy of the NF1 gene in their body, but only develop tumors when both copies are nonfunctional in glial cells, Nf1+/− mice were generated that lack Nf1 inactivation in astrocytes. The Nf1 conditional knockout mice develop gliomas in the prechiasmatic optic nerves and chiasm (8). Optic gliomas form in these mice by 10–12 weeks of age (60). Gross inspection of the optic nerves in these mice reveals bilateral prechiasmal optic nerve and chiasm enlargement (Fig. 4b), similar to what is observed in children with NF1. Microscopically, the areas of optic nerve enlargement are accompanied by increased numbers of GFAP-positive astrocytes, elevated proliferative indices, and cells with atypical, hyperchromatic nuclei. In contrast to their human counterparts, however, the Nf1 GEM gliomas lack microcystic spaces, Rosenthal fibers, and granular bodies (8).
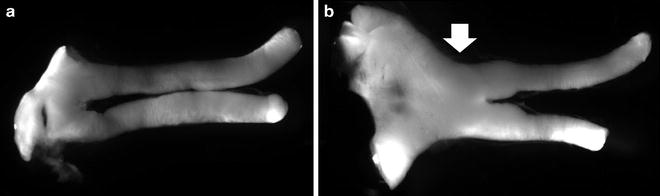
Fig. 4.
Representative optic nerves and optic glioma from Nf1 genetically engineered mice. (a) The normal optic nerve and chiasm from a representative control mouse is shown. (b) A representative optic glioma in a genetically engineered mouse is demonstrated by the presence of swollen optic nerves and chiasm.
The Nf1 optic glioma GEM model has been employed in preclinical studies to demonstrate that conventional single-agent chemotherapy (temozolomide) reduces glioma size and proliferation (50). In addition, biologically based therapies, including rapamycin, have been tested for efficacy in this model, and the positive results were used to propel clinical trials in children with NF1-associated brain tumors.
3.4 The Role of the Tumor Microenvironment in Glioma Growth
NF1-associated PAs have a predilection for the optic pathway, which prompted a closer investigation of the nonneoplastic cell types and growth factors in this brain region. Endothelial cells and microglia, macrophage-like immune system cells, are among the noncancerous cells that produce growth factors that increase preneoplastic and neoplastic cell growth (61). Microglia with reduced Nf1 gene expression (Nf1+/− microglia) are found in increased numbers prior to obvious optic glioma formation in Nf1 optic glioma mice (60). Importantly, inhibition of microglial function attenuates optic glioma proliferation in vivo (62, 63). These findings underscore the importance of microglia and nonneoplastic cells in the tumor microenvironment to optic glioma growth, and present another target for future glioma therapies.
3.5 Visual Impairment Following Optic Glioma Formation
Nf1 optic glioma mice have been used to understand the cellular and molecular basis for visual loss in children with NF1-associated optic glioma. Visual impairment in children with NF1-associated OPG is frequently encountered, with only 20% of children exhibiting improvements in vision following chemotherapy (45). Nf1 optic glioma mice exhibit reduced visual evoked potentials during early tumor formation, suggesting abnormal neuronal transmission (64). Moreover, there is evidence of axonal deterioration and increased retinal ganglion cell (RGC) apoptosis in response to glioma formation and progression (64–66). To gain insights into the impact of reduced Nf1 gene expression on RGC function, Nf1+/− RGCs were grown in vitro. Surprisingly, Nf1+/− RGCs had a cell-autonomous decrease in neurite lengths, growth cones, and survival (56). These Nf1+/− RGC abnormalities were the result of reduced neurofibromin-regulated cAMP levels and could be rescued by restoring cAMP levels in vitro (56, 67). Relevant to visual impairment in children with NF1-associated optic glioma, pharmacological restoration of cAMP levels with Rolipram partly restored RGC survival in vivo (56), suggesting that future treatments for symptomatic NF1-associated OPG might involve neuroprotective strategies.
3.6 Genomic Influences on Nf1 GEM Tumorigenesis
Since the majority of children with NF1 do not develop brain tumors, recent studies have explored the influences of genomic variation on glioma susceptibility. Nf1 GEM mice heterozygous for inactivating mutations involving both the Nf1 and Trp53 genes (NPCis mice) are predisposed to the development of high-grade astrocytomas (68, 69). NPCis mice maintained on the C57Bl/6 genetic background develop high-grade astrocytomas, whereas NPCis mice maintained on the 129 S4 genetic background are relatively resistant to astrocytoma formation (70). These findings suggest that genomic modifiers involving genes other than the NF1 gene may influence glioma penetrance. The identification of these modifier loci in GEM models coupled with genome-wide association studies in children with NF1 could lead to the early identification of children most at risk for developing a brain tumor.
4 Conclusion
Nf1 GEM glioma models have greatly expanded our understanding of the cellular, molecular, and genomic basis for brain tumor development. These models have identified critical signaling pathways responsible for astrocyte proliferation and survival. Furthermore, these models have helped researchers understand how the nonneoplastic tumor microenvironment can dictate where and when brain tumors form, and how these nonneoplastic cells can be targeted to reduce glioma growth. The availability of robust small-animal models of NF1-associated glioma provides unique opportunities to develop treatments that target both neoplastic and nonneoplastic cells as well as protect neurons from damage. Importantly, these GEM models establish preclinical platforms for the rapid evaluation of promising therapies prior to their use in children with NF1. With continued progress in genomics and mouse modeling, we are nearing an era in which genetic information may identify at-risk individuals and facilitate the development of “personalized” treatment regimens.
Acknowledgments
We thank Dr. Sonika Dahiya and Mr. Scott Gianino for the images of human pilocytic astrocytoma specimens and mouse Nf1 optic gliomas, respectively. Ibrahim Hussain is supported by funding from the Doris Duke Charitable Foundation to Washington University School of Medicine. David H. Gutmann is supported by funding from the National Cancer Institute, the National Institutes of Health, the James S. McDonnell Foundation, and the Department of Defense.
References
1.
2.
Bar EE, Lin A, Tihan T, Burger PC, Eberhart CG (2008) Frequent gains at chromosome 7q34 involving BRAF in pilocytic astrocytoma. J Neuropathol Exp Neurol 67:878–887PubMedCrossRef
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


