Chapter 4 General Pathology of the Eye
Ocular pathology is relevant to those interested in clinical veterinary ophthalmology because examination of the eye is the direct observation of its gross pathology; even the ophthalmoscope is a low-power microscope. There is a strong correlation between the observations made on clinical examination and the results of microscopic examination. Clinicians and pathologists use exactly the same terminology—a welcome accord that is almost unique among medical specialties and is further testimony to the inseparability of clinical ophthalmology and ophthalmic pathology.
OCULAR INJURY
Consequences of Ocular Injury
At the cellular level the reaction of ocular tissue to injury is the same as elsewhere, and depends on the nature, duration, and severity of the insult. The response to the injurious stimulus is one or more of the following: resistance, adaptation, injury, containment, and repair.
Hypertrophy
Hypertrophy is defined as an increase in tissue mass because of an increase in cell size. It is most commonly encountered as hypertrophy of the retinal pigment epithelium (RPE) subsequent to retinal detachment of more than a few hours’ duration. It is a rapid, reliable, and more or less specific change that allows one to separate genuine detachment from artifact (Figure 4-1). Hypertrophy is also seen in many other ocular tissues as a prelude to replication, such as occurs with hypertrophy of iris endothelium preparatory to the development of a preiridal fibrovascular membrane, hypertrophy of corneal stromal fibroblasts in the early stages of stromal repair, and hypertrophy of lens epithelium at various stages of cataract formation.
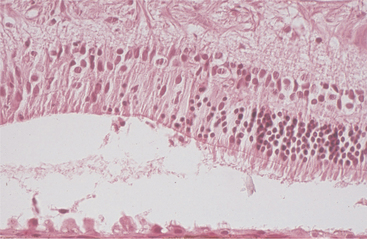
Hyperplasia
Hyperplasia is defined as an increase in tissue mass because of an increased number of cells. It may or may not be accompanied by hypertrophy of the same cells. In general, hyperplasia is the more efficient response to a need for more tissue mass in all tissues that are capable of mitotic replication. It is seen under many of the same circumstances as hypertrophy, and is relatively less common in the eye than it is in most other tissues because so many of the ocular tissues have limited or no proliferative capability. Many of the most dramatic examples of hyperplasia within the eye are combined with metaplastic changes, as described later. In most instances hyperplasia within the eye is seen as a transient phase of tissue regeneration that precedes eventual normalization of the reparative tissue mass. It is seen in conjunctival and corneal epithelium following any kind of injury and may remain in a permanently excessive state in the form of an epithelial facet (a plaquelike corneal epithelial thickening). Similarly, it may remain as a plaque of hyperplastic lens capsule epithelium in the form of an anterior capsular or subcapsular cataract (Figure 4-2).
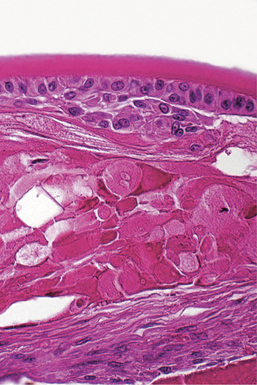
Atrophy
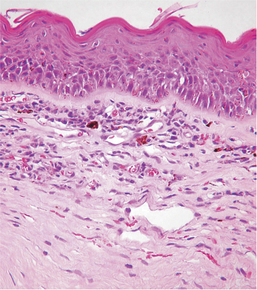
Atrophy implies the reduction of tissue mass at some point after the tissue had reached its full development. Causes are as diverse as ischemia, denervation, lack of hormonal or other trophic stimulation, disuse, and loss of mass due to degeneration or necrosis. Within the eye “atrophy” is used most frequently to describe senile iris atrophy and pressure-induced glaucomatous atrophy of ciliary processes, retina, and optic nerve, and as a relatively imprecise umbrella for a group of so-called retinal atrophies (Figures 4-3 and 4-4).
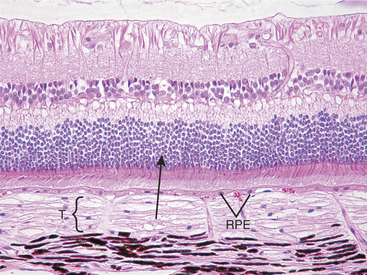
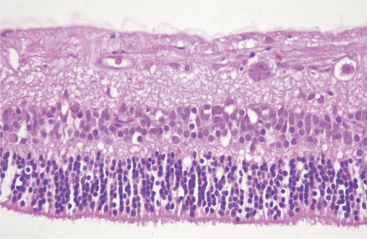
Metaplasia
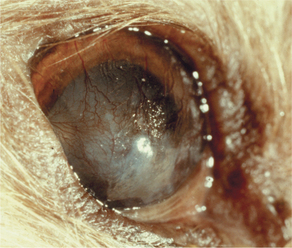
Metaplasia is the conversion of one adult tissue type into another, related and more durable, tissue type. The most prevalent examples are conversion of fibrous tissue into bone, or columnar mucosal epithelium into stratified squamous epithelium. The usual stimulus seems to be the need to adapt to a more hostile environment by acquiring a more durable cellular phenotype. Metaplasia is a relatively uncommon reaction in most tissues, but it is a particularly prevalent and clinically important reaction within the eye. Common examples are so-called cutaneous metaplasia of the cornea in cases of chronic keratoconjunctivitis sicca or exposure keratitis, under which circumstances the cornea seems to recall its embryologic origins as skin, and thus undergoes keratinization, pigmentation, and vascularization (Figures 4-5 and 4-6). In this instance metaplasia is a protective adaptation, because the epithelium shifts to a phenotype more able to withstand dryness or chronic abrasion. Similarly, conjunctiva commonly undergoes metaplasia to a stratified squamous (and sometimes keratinized) epithelium in response to chronic irritation.
Dysplasia
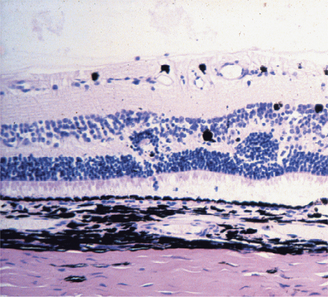
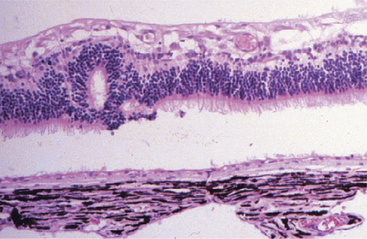
Dysplasia means disorderly proliferation. The term can be used in an embryologic context to signify disorderly development of a tissue, in the context of wound healing to imply a transient jumbling of tissue organization that precedes normalization, or in a neoplastic context to describe the state of disordered proliferation that is a prelude to malignant transformation. In ocular pathology we use it in all three contexts. Familiar examples are jumbled retinal development that may be inherited or may follow viral or chemical injury to the developing retina (Figures 4-7 and 4-8), conjunctival or corneal epithelial dysplasia that is a prelude to actinic squamous cell carcinoma, and nonneoplastic jumbling of corneal epithelium in any healing corneal ulcer (see Figure 4-5).
Necrosis
LOSS OF BARRIER FUNCTION.
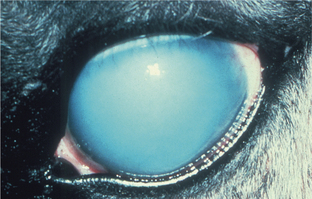
Normal ocular function depends on the preservation of numerous critical barriers to maintain ocular clarity. For example, we ordinarily detect corneal epithelial necrosis only because the underlying hydrophobic collagenous stroma osmotically imbibes the water from the tear film, resulting in rapid localized corneal opacity (Figures 4-9 and 4-10). Staining of the defect by fluorescein to facilitate clinical detection operates on exactly the same principle, as this hydroscopic dye binds to the now-exposed collagenous stroma. Similarly, necrosis of corneal endothelium results in deep corneal stromal edema, and necrosis of lens epithelium results in hydropic swelling of the normally dehydrated lens fibers, which we detect as cataract (see Figure 4-2). Loss of the endothelial tight junctions (as in vascular hypertension, infection with such organisms as the rickettsiae of Rocky Mountain spotted fever) within the iris or choroidal blood vessels results in serous effusion or even hemorrhage, which interferes with the clarity of the ocular media, or more serious consequences of retinal ischemia.

OCULAR INFLAMMATION
Philosophically, inflammation should be considered a transient, controlled vascular and cellular response by living tissue to injury of almost any type. That response should be qualitatively and quantitatively appropriate to the nature of the tissue injury, and it should serve to neutralize and remove the injury’s stimulus while also laying the groundwork for parenchymal and stromal repair. In evolution, inflammation probably first appeared as a dèbridement phenomenon, whereby macrophages or their equivalent would move into injured tissue, clear up the debris, and prepare the way for healing to occur. We now know that this dèbridement itself consists of at least two simultaneous phenomena, namely, the removal of tissue debris and the production of various growth factors that initiate the subsequent tissue repair. Later in evolution the inflammatory process assumed a more defensive role, tightly integrating with the immune system to recognize and destroy a variety of infectious and noninfectious “foreign” agents.
The leukocytes that thus enter the tissue become intermingled with the serum and fibrin that may already have accumulated because of the previous increase in vascular permeability, and these mixtures form the inflammatory exudates that for years have formed the basis for the prediction of disease causation based on histologic or cytologic evaluation of such exudates. These exudates are not static but are constantly changing in amount and in cellular and humoral makeup according to the ever-changing nature of the battle between the injurious agent and the tissue defenses. In its simplest form these changes may be nothing more than a gradual reduction in the intensity of the vascular response and leukocytic recruitment as the humoral and cellular defenses accomplish their task of diluting and destroying the offending agent. On the other hand, the nonspecific humoral and cellular defenses may become modified by the addition of specific immune responses as the battle continues. These immune responses act, in general, to improve both the specificity and the efficacy of what at first are relatively broad and nonselective defensive strategies.
Peculiarities of Ocular Inflammation
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


