CHAPTER 20 Equine Alphaviruses
The first recorded epidemic of eastern equine encephalitis (EEE) in horses likely occurred in Massachusetts in 1831; the first recorded human case occurred in that state in 1838.1 In 1933, EEE virus was isolated from a horse, and it was established that epidemics of encephalitis in horses in North America were caused by two separate viruses, EEE and western equine encephalitis (WEE), that were segregated geographically. A related virus, Venezuelan equine encephalitis (VEE), causes outbreaks of encephalitis in horses in Central America, South America, Mexico, and occasionally the southern United States. Although widespread vaccination has reduced the size and number of outbreaks of EEE, WEE, and VEE in horses, the impact of these diseases is still significant because of the fulminant nature of clinical signs and high mortality rate in affected horses.
ETIOLOGY
The genus Alphavirus belongs to the family Togaviridae and includes a large number of viruses that have been isolated from horses with neurologic disease. Of these alphaviruses, eastern, western, and Venezuelan equine encephalitis viruses are the most frequently isolated from epidemics of encephalitis in horses and humans in the Western Hemisphere.2,3 The other genus in the family Togaviridae, Rubivirus, contains no viruses of known equine significance. Togaviruses are single-stranded, linear positive-sense ribonucleic acid (RNA) viruses that are enveloped and measure 60 to 70 nm in diameter. Within the envelope there is a nucleocapsid with icosahedral symmetry composed of peplomers arranged as trimers. Each peplomer is a heterodimer composed of two glycoproteins, E1 and E2.4
The glycoproteins E1 and E2 are immunodominant proteins that induce neutralizing antibody.4–6 The E2 glycoprotein induces the strongest neutralizing antibody response (both polyclonal and monoclonal) and has hemagglutinating properties, the activity of which is highly modulated by pH.2,7,8 Both hemagglutination inhibition (HI) activity and neutralizing specificity have historically been used to differentiate viral species and their antigenic types (Table 20-1). Although these techniques are now being rapidly replaced by molecular sequencing as the basis for viral classification, the common group-specific antigenic determinants are still usually defined by serologic techniques, such as fluorescent antibody (FA), complement fixation (CF), and enzyme-linked immunosorbent assay (ELISA).7–15
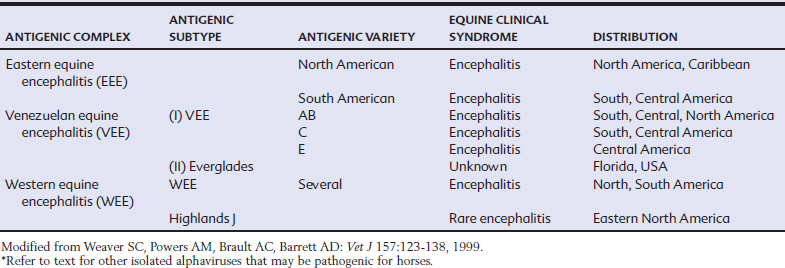
In addition to the alphaviruses that occur in the Western Hemisphere and are associated with equine encephalitis, several other alphaviruses that are potential causes of encephalitis have been isolated in the Americas and throughout the world.16 There is only one known species of EEE virus, but it exists as separate North and South American variants with a high degree of genetic conservation between isolates.17 North American isolates of EEE virus differ by less than 2% in their genomic nucleotide sequence analysis, even when comparing strains isolated more than half a century apart and over a 1200-mile land mass. In contrast, South American isolates can be classified as several genotypes that diverge by up to 25% in nucleotide sequence analysis.18
There are two antigenic subtypes of WEE virus: WEE and Highlands J viruses.19–23 Most infections that occur east of the Mississippi River are caused by the Highlands J (HJ) virus.20,24–26 Although generally considered less pathogenic for mammals than WEE virus, HJ virus has caused natural cases of encephalitis in horses and is pathogenic for domestic turkeys, pheasants, and exotic species of birds.20,24,25 Other subtypes of WEE virus that may cause disease in North American horses include Fort Morgan and Buggy Creek viruses.27 One WEE variant, Y62-WEE, has been identified in Russia. No equine disease has been associated with either Sindbis or Aura viruses, other members of the Alphavirus family.28 Most WEE infections in the western United States are likely the result of infection with one of the several antigenic variants of the actual WEE subtype.29 This virus is more pathogenic in horses and humans than closely related viruses, although minimal disease has been reported in humans and horses in the last decade.
Six distinct subtypes (designated by Roman numerals I through VI) and numerous varieties of viruses (designated by letter) within those subtypes are classified within the VEE virus complex.9 The “epidemic type” of VEE viruses (types IAB, IC, and IE) are responsible for the large outbreaks of encephalitis in horses in the Western Hemisphere in the past 20 years. So-called endemic types of VEE virus are considered to be of low pathogenicity for equids under most circumstances.29–31 These include ID and IF variants from Central America and Brazil, respectively,9,32 type II (Everglades) virus found in Florida, three known variants of type III (Mucambo) virus, type IV (Pixuna) virus, type V (Cabassou) virus, and type VI virus. The Mucambo virus has three subtypes of potential pathogenicity for the horse.9,33 These viruses have been isolated in Trinidad, French Guiana, western North America, and Peru. The Pixuna subtype of VEE is associated with febrile illness in horses in Brazil.9
EPIDEMIOLOGY
The life cycle of equine alphaviruses involves transmission between birds or rodents and mosquitoes.34 In some cases, other domestic and wild animal species, especially species exotic to North America, such as the emu, have been affected during these outbreaks.35 Although other RNA viruses, such as human immunodeficiency virus (HIV), have high rates of genetic evolution (up to 104 nucleotide changes per year), alphaviruses evolve relatively slowly, at approximately 102 to 103 substitutions per year.22,36–39 This slow progression reflects adaptation of viruses to multiple hosts, which presumably requires more genomic conservation for maintenance in nature. There may be higher rates of viral evolution outside of North America. Despite this lower evolution rate, these viruses have the ability to adapt rapidly to new ecologic challenges. When VEE is placed in cell culture, serial passage results in stability of the substitution rate. When placed back in vivo (usually hamsters), the mutation rate of the virus increases dramatically, indicating that these viruses can rapidly change and have the potential to cause new outbreaks in new hosts and in new locales. Because alphaviruses are transmitted by arthropod vectors, clinical disease occurs during the arbovirus season of late summer and early fall in temperate zones, with year-round transmission possible in the tropics and subtropics.
Understanding the antigenic and genetic relationships among the viruses in the WEE complex has proved more challenging than for the viruses within the EEE and VEE complexes. Western equine encephalitis virus is a member of the WEE antigenic complex that includes several Old World viruses in addition to the New World viruses previously described. Phylogenetic analyses of isolates from North and South America indicate that regional WEE lineages appear to have evolved independently for several years to a few decades (e.g., genotypes in South America are apparently absent from North America).3,25,37,40–44 However, relative homogenous genotypes of WEE are dispersed across both North and South America. This contrasts with EEE and VEE viruses, where certain virus genotypes appear to be restricted to either North or South America. WEE virus has been reported in several countries in South America (Argentina, Guyana, Ecuador, Brazil, Uruguay), but only in Argentina has it been associated with human disease and significant epidemics in horses.23,45–47
Eastern Equine Encephalitis
Although designated as an “eastern” virus, EEE has a wide geographic distribution. It is found as far north as eastern Canada, is dispersed throughout the Caribbean, and has been identified in Central and South America. Infection in the United States (U.S.) is primarily seen in the southeastern states but has been detected in all states east of the Mississippi River and also in a number of western states. In recent years, intense focal activity has been reported in Wisconsin, Ohio, Massachusetts, and New Hampshire.48,49
In North America, EEE virus is perpetuated in a sylvatic cycle between avian hosts (passerine birds) and mosquitoes, primarily the ornithophilic mosquito, Culiseta melanura.50–52 Indigenous passerine birds do not develop disease but develop sufficient titer viremia to allow transmission to feeding mosquitoes.53 Cs. melanura is not a mammalian feeder and is not responsible for transmission between birds and mammalian hosts. However, several species of secondary or epidemic mosquitoes that feed on both birds and mammals can act as biologic vectors. These likely transmit EEE to horses, humans, and other vertebrate species. Horses and humans are clinically affected but do not develop viremia sufficiently high enough to transmit virus back to vector mosquitoes and are considered “dead-end” hosts.
Disease caused by EEE was first identified in Connecticut in 1937 in the then recently introduced exotic ring-necked pheasant. Since that time, disease in sparrows, pigeons, Pekin ducks, and Chukar partridges (all old-world species) has been reported.54 In 1991, EEE was the cause of fatal hemorrhagic colitis in commercial flocks of emus in Louisiana. Similar outbreaks have been reported in emus and ostriches in Georgia, Florida, and other states.55,56 Outbreaks of EEE have been recorded in intensive swine herds in southern Georgia.57 Isolated cases of EEE in cattle, sheep, and nondomestic ungulates have also been recorded.58–60
The mechanism by which EEE virus overwinters (diapause) remains uncertain.61,62 Transovarial transmission of EEE virus is not important, although it does occur in the mosquito host. In tropical and subtropical climates, the year-round mosquito/avian cycle likely obviates the need for a period of diapause in mosquito populations. In the many areas of North America with seasonal EEE activity, winters are long and severe enough such that no adult mosquito activity occurs for several months.
Culiseta melanura, a temperate breeding species of mosquito, does not readily breed in southern Florida and the Caribbean, and EEE is not an endemic disease in this relatively focal region.63 Only sporadic reports or small epidemics of EEE disease in horses have been recorded in these areas, likely through migratory influx of viremic birds providing occasional sources of virus for secondary vectors. These secondary vectors can initiate short-term outbreaks but cannot maintain the disease endemically.
Comparatively few epidemics of EEE in horses have occurred in South America, with minimal disease reported in humans. Mosquitoes of the subgenus Culex (Melanoconion) in South and Central America are implicated as endemic vectors.64 Antibody prevalence studies in birds and small rodents indicate that, in contrast with North America, small rodents are involved in the primary virus life cycle.
Western Equine Encephalitis
Since 1964, WEE has been reported in 640 people. The highest number of cases has been reported in Colorado and Texas, followed by Minnesota and California. Historically, large outbreaks of WEE have been described in horses. The virus was first identified in association with a large epizootic that occurred in the San Joaquin Valley of California in 1930. Approximately 6000 horses were affected, with a case-fatality rate of 50%.65 This outbreak continued and spread to several western states from 1931 to 1934. Within a decade, another 300,000 equid cases were reported, with several thousand human cases. Over the last decade, reports of WEE in horses have been limited and sporadic, likely reflecting vaccination and protective immunity gained by subclinical exposure.66,67
Culex tarsalis is the primary vector that maintains WEE virus in an enzootic cycle with birds, especially nestling passerines,68,69 Cx. tarsalis population abundance is favored by a rapid increase in temperature following a cool, wet spring, resulting in the rapid melting of snow and flooding of rivers.70,71 This species of mosquito also has a predilection for irrigated lands as breeding sites.72 Other ornithophilic mosquitoes become infected as the summer progresses, and the infection eventually spills over to other types of birds, mammals, and possibly reptiles and amphibians. Most, if not all, of these infections are inapparent. This in turn results in the virus becoming established in species of mosquitoes with host preferences other than birds. Transovarial transmission of WEE in North American mosquitoes, including Cx. tarsalis, has not been proved, and interepidemic maintenance in temperate areas by mosquitoes is therefore in doubt. Horses, even those that are obviously clinically affected, do not produce viremia high enough to infect mosquitoes.
At least two variants of WEE virus (Fort Morgan and Buggy Creek) have been isolated in western North America and are transmitted between birds by swallow nest bugs (Oeciacus vicarius).24 Neither variant is considered to be pathogenic for humans or horses. A third variant, Highlands J virus, is mainly found east of the Mississippi River and has been isolated from horses dying of encephalitis.20,24–26,73 Information is limited on the number of horses infected with HJ virus on an annual basis.
In South America, where Cx. tarsalis does not occur, antibody prevalence rates in birds are lower than in North America, and the species (vertebrate and invertebrate) responsible for maintaining the virus on that continent have not been identified.74 Aedes albasfaciatus has experimentally transmitted WEE virus to chickens in Argentina.75 Other studies have demonstrated mosquitoes of the Culex pipiens complex are refractory to oral WEE virus inoculation. The species of mosquitoes from which WEE virus has mostly been isolated feeds principally on mammals rather than birds.
Venezuelan Equine Encephalitis
Venezuelan equine encephalitis virus is one of the most important human and veterinary pathogens in the New World.76 The virus, both historically and very recently, has been responsible for large outbreaks of disease in both humans and horses over large geographic areas. The first recognized outbreak of VEE occurred initially in equids in Colombia and then in Venezuela in 1935 and 1936, although it is speculated to have been active in this area since 1920.77 Documentation of human disease occurred in a Columbian outbreak in the 1960s, when an estimated 50,000 to 100,000 equids (horses, mules, donkeys) died and 250,000 humans were affected (mainly an influenza-like disease, but some cases of encephalitis and death). It is uncertain whether the 1969–1971 epidemic that was first reported in Ecuador and subsequently spread to Central America, Mexico, and Texas was directly related to this outbreak in Colombia or was caused by the use of an incorrectly inactivated subtype IAB strain vaccine.78,79 Regardless of its cause, however, this epidemic revealed the potential for VEE to spread rapidly within an equine population, with a case-fatality rate approaching 90% in some areas.
The availability of vaccines and active surveillance throughout the Americas since the early 1970s have arguably reduced the impact of the disease in the rural regions, where VEE has traditionally been described. Nevertheless, after a long period with no evidence of clinical disease in horses, outbreaks of VEE were reported in 1993 in Chiapas, southern Mexico; in 1995 in Venezuela and Colombia; and in 1996 in Oaxaca, Mexico.80–84 The geographically extensive outbreak in 1995 had all the initial hallmarks of the 1969–1971 epidemic. Not only did large numbers of horses, mules, and donkeys die, but there were also an estimated 75,000 to 100,000 human cases of disease. VEE viruses isolated in 1995 were genetically similar to those associated with disease in the 1960s. Although the cause of these severe cyclic disease occurrences is still a matter of intense research, it can be assumed that severe epizootic VEE may continue to occur with approximately one- to two-decade-long interepizootic periods.
Key to understanding the epidemiology of VEE is recognition of the differences in the basic biology of two transmission cycles, enzootic and epizootic, of this virus76 (Fig. 20-1). The enzootic cycle centers around sylvatic rodents such as spiny and cotton rats, which have high natural infection rates and can develop viremia high enough to transmit VEE to mosquitoes.85,86 Even opossums, bats, and shore birds likely are important in dispersal of enzootic virus.87,88 The subgenus Melanoconion (Culex cedecci) is likely to be the most important vector of enzootic VEE.89 This vector resides in tropical forests and swamps and feeds on small forest mammals at night. Some species of this mosquito have broader feeding patterns. Activity for this vector peaks with high ambient temperature and rainfall. Table 20-1 indicates that certain strains of virus are found only in the enzootic cycle. These viruses, subtypes I-E, II, III, and IV, tend to be of low pathogenicity for equids and do not result in high levels of viremia in horses.
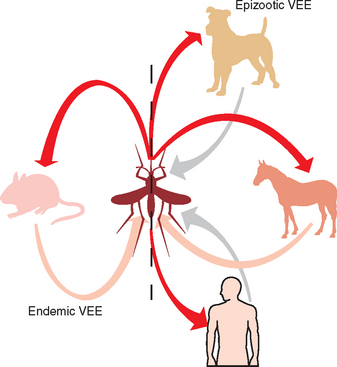
Many theories exist on the origin of epizootic VEE viruses, primarily of the subtypes IAB and IC.90,91 These viruses are associated with variable but often quite high equine mortality (20%-85%).92 In contrast to most arboviruses in the horse, efficient amplification of the virus by equids is the hallmark of epizootic VEE. Humans usually develop a flulike illness, with only 4% to 14% exhibiting neurologic signs and symptoms.88 Case-fatality rate for humans is approximately 1%. Several other species of mammals, including domestic rabbits, small ruminants, and dogs, develop potentially fatal clinical disease after VEE virus infection.92 More than 100 species of birds have been either virologically or serologically associated with transmission of epidemic VEE virus. Shore birds in general and herons in particular appear to be capable of serving as amplifier hosts.93,94 Birds may develop viremia as high as 108 TCID50/mL of blood.
The importance of equine infection in maintenance of epizootic VEE is evidenced by the observation that human disease has never been demonstrated in the absence of equine disease.29 All mammalian hosts are capable of developing a high-titer viremia of approximately 105 to 107 pfu/mL for up to 5 days, but the horse is likely to be the most important mammalian host in terms of vector capacity.76 In contrast to EEE and WEE, where horses are not considered to be a major source of virus for the vector, in VEE epidemics, horses are the most important amplifiers of virus activity.
Several species of mosquitoes from at least 11 genera have been determined to be naturally infected with epidemic strains of VEE virus.95–102 In particular, Psorophora confinnis, P. columbiae, Ochlerotatus sollicitans, O. taeniorhynchus, and Culex spp. have been associated with epizootics. Virus has also been isolated from Culicoides spp. (Ceratopogonidae) and blackflies (Simuliidae), but it is not known whether insects in these families are capable of biologic transmission of VEE virus.95 During an epidemic, dogs regularly become infected and may be capable of virus amplification.103–106 In addition, ticks, including the species Ambylomma cajennense and Hyalomma truncatum, may be capable of viral transmission.107–111
Several theories exist regarding the source of IAB and IC strains and how they persist in the environment between outbreaks.112 Although molecular analyses have been used to address this, a comprehensive understanding of how epidemics in horses originate remains elusive. Isolates that are virulent for horses do not appear to be transmitted in the interepizootic period.113,114 There is some evidence for circulation of these viruses in the interepizootic period, but no latency is associated with these infections. No evidence suggests that the epizootic strains coexist in the enzootic cycle. Mutation of enzootic strains may allow the emergence of highly pathogenic virus and initiation of epizootics.115 This has been identified as the source of four epizootics. Some of these epizootics may have occurred secondary to the use of a modified live vaccine derived from the IAB strain.79
PATHOGENESIS
The alphavirus genome is 9.7 to 11.8 kilobases in length and encodes both nonstructural and structural proteins. With a 5′-methylated and a 3′-polyadenylated cap, the nonstructural proteins are encoded at the 5′ end and the structural proteins at the 3′ end of the genome.40,116–119 Unlike flaviviruses, which translate the entire genome, only the 5′ end is translated in alphaviruses. The resultant polyprotein is subsequently cleaved by a proteinase. Viral RNA-dependent RNA polymerase is formed from two of these proteins, and complementary (negative-sense) RNA (cRNA) is transcribed.3,120 From this template, full-length viral progeny are transcribed, and a short “subgenomic” portion is transcribed.121–123 The latter, which has a cap at the 5′ end and a polyadenylated tail, is translated to form a polyprotein that is processed to the five viral proteins: E1, E2, E3, 6K, and C.117,119,124–127
Alphaviruses replicate to high titer in the cytoplasm of infected cells and exit the cell by the budding of preassembled nucleocapsids through the plasmalemma.128,129
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


