Chapter 180 Diuretics
PHARMACOLOGY
Diuretics are grouped according to their mechanism of action and they include, in order of their renal tubular target, osmotic diuretics, carbonic anhydrase (CA) inhibitors, loop diuretics, thiazide diuretics, aldosterone antagonists, and other potassium-sparing distal diuretics (Table 180-1). Mannitol and furosemide are used most frequently in the critical care setting; consequently the other diuretics will be mentioned here only briefly. Usual dosage recommendations are summarized in Table 180-2. In using diuretics it is important to note that fluid therapy should be adjusted closely to the desired goals of the global therapy. For example, if the therapeutic goal is a depletion of the ECF with furosemide in an animal with CHF, it does not make sense to administer intravenous fluids concomitantly. Partial free water replacement with 5% dextrose in water may be an exception in this scenario.
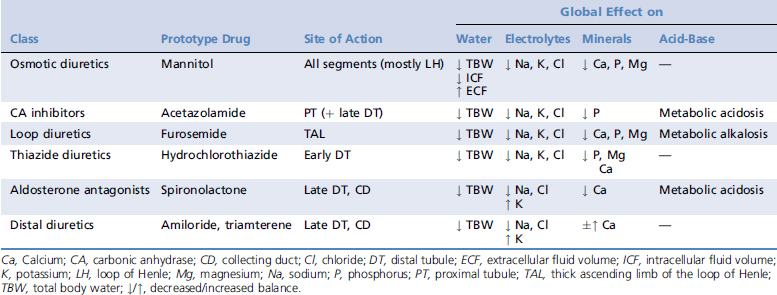
Table 180-2 Dosages of Most Commonly Used Diuretics
| Drug | Indication | Dosage |
|---|---|---|
| Mannitol | Renal failure | 0.25 to 1 g/kg IV q4-6h CRI 1 to 2 mg/kg/min when diuresis instituted |
| Glaucoma | 1 to 3 g/kg IV once | |
| Cerebral edema | 1 to 1.5 g/kg IV once | |
| Acetazolamide | Glaucoma | 50 mg IV once, then 2 to 10 mg/kg q8-12h PO 7 mg/kg PO q8h in the cat |
| Furosemide | Diuretic | 0.5 to 4 (max 8) mg/kg IV, IM, SC, PO q8-12h CRI 2 to 15 μg/kg/min |
| Hydrochlorothiazide | Diuretic | 0.5 to 5 mg/kg PO q12-24h |
| Spironolactone | K-sparing diuretic | 1 to 4 mg/kg PO q12-24h |
| Amiloride | K-sparing diuretic | 0.1 to 0.3 mg/kg PO q24h |
| Triamterene | K-sparing diuretic | 1 to 2 mg/kg PO q12h |
CRI, Constant rate infusion; IM, intramuscular; IV, intravenous; K, potassium; PO, per os; SC, subcutaneous.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


