Chapter 3 Disorders of Sodium and Water
Hypernatremia and Hyponatremia
The volume and tonicity of body fluids are maintained within a narrow normal range by regulation of sodium and water balance. The volume of extracellular fluid (ECF) is determined by the total body sodium content, whereas the osmolality and sodium concentration of ECF are determined by water balance. The kidneys play a crucial role in these processes by balancing the excretion of salt and water with their intake and by avidly conserving them when intake is restricted (Table 3-1).
Table 3-1 Renal Regulation of Sodium and Water Balance
| Osmoregulation | Volume Regulation | |
|---|---|---|
| What is sensed | Plasma osmolality | Effective circulating volume |
| Sensors | Hypothalamic osmoreceptors | Carotid sinus |
| Aortic arch | ||
| Glomerular afferent arterioles | ||
| Cardiac atria | ||
| Large pulmonary vessels | ||
| Effectors | Vasopressin | Renin-angiotensin-aldosterone system |
| Thirst | Sympathetic nervous system | |
| Atrial natriuretic peptide | ||
| “Pressure natriuresis” | ||
| Antidiuretic hormone | ||
| What is affected | Water excretion | Urine sodium excretion |
| Water intake |
Modified from Rose BD. Clinical physiology of acid base and electrolyte disorders, 4th ed. New York: McGraw-Hill, 1994: 256, with permission of the McGraw-Hill Companies.
Terminology
Osmolality
The normal plasma osmolality of dogs and cats is slightly higher than that of humans and ranges from 290 to 310 mOsm/kg in dogs and from 290 to 330 mOsm/kg in cats. In one study, 20 dogs under resting conditions had plasma osmolality values of 292 to 308 mOsm/kg with a mean value of 301 mOsm/kg.67 In a study of the effects of sodium bicarbonate infusion in cats, baseline serum osmolality ranged from 290 to 330 mOsm/kg.22 Plasma osmolality can be estimated from the equation:
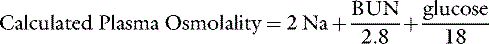
where BUN is blood urea nitrogen. In this equation, the concentrations of urea and glucose in milligrams per deciliter are converted to millimoles per liter by the conversion factors 2.8 and 18. The measured osmolality should not exceed the calculated osmolality by more than 10 mOsm/kg.42,149 If it does, an abnormal osmolal gap is said to be present. This occurs when an unmeasured solute (i.e., one not accounted for in the equation) is present in large quantity in plasma (e.g., mannitol or metabolites of ethylene glycol) or when hyperlipemia or hyperproteinemia results in pseudohyponatremia (see section on Hyponatremia with Normal Plasma Osmolality).42,50,56
Specific gravity
The term specific gravity refers to the ratio of the weight of a volume of liquid to the weight of an equal volume of distilled water. Specific gravity depends not only on the number of particles present in the solution but also on their molecular weight. The clinician can easily measure specific gravity with a hand-held refractometer. Multiplying the last two digits of the urine specific gravity (USG) by 36 gives a rough estimate of urine osmolality in dogs.71 This rule may be misleading if the urine sample contains a large amount of high-molecular-weight solute, because substances with high molecular weights have a greater effect on specific gravity than on osmolality. The effects on urine osmolality of some solutes are shown in Table 3-2.
Table 3-2 Effect of Selected Solutes on Urine Osmolality⁎
| Substance | Molecular Mass (da) | Contribution to Osmolality (mOsm/kg) |
|---|---|---|
| Albumin | 69,000 | 0.144 |
| Diatrizoate ion | 613 | 16.313 |
| Glucose | 180 | 55.555 |
⁎ 1.0 g/dL of each of the listed solutes added to distilled water would increase specific gravity by 0.010, but would have the effects on osmolality shown in the table.
Tonicity or effective osmolality
Changes in the osmolality of ECF may or may not initiate movement of water between the intracellular and extracellular compartments. A change in the concentration of permeant solutes (e.g., urea, ethanol) does not cause movement of water because these solutes are distributed equally throughout total body water (TBW). A change in the concentration of impermeant solutes (e.g., glucose, sodium) does cause movement of water because such solutes do not readily cross cell membranes. Tonicity refers to the ability of a solution to initiate water movement and is dependent on the presence of impermeant solutes in the solution.41 Thus, tonicity may be thought of as effective osmolality. A solution is hypertonic to a reference solution from which it is separated by a semipermeable membrane if its concentration of impermeant solutes is greater than that of the reference solution. A solution is hypotonic to the reference solution if its concentration of impermeant solutes is less than that of the reference solution. A solution is isotonic to the reference solution if its concentration of impermeant solutes equals that of the reference solution.
Types of dehydration
Dehydration occurs when fluid loss from the body exceeds fluid intake. Dehydration may be classified according to the type of fluid lost from the body and the tonicity of the remaining body fluids. Pure water loss and loss of hypotonic fluid result in hypertonic dehydration because the tonicity of the remaining body fluids is increased. Loss of fluid with the same osmolality as that of ECF results in isotonic dehydration, because there is no osmotic stimulus for water movement and the remaining body fluids are unchanged in tonicity. Loss of hypertonic fluid or loss of isotonic fluid with water replacement results in hypotonic dehydration because the remaining body fluids become hypotonic. The types of dehydration and their relative effects on the volume and tonicity of the intracellular and extracellular compartments are shown in Figure 3-1.

Normal physiology
Renal handling of sodium
Approximately 25% of the filtered load of sodium is reabsorbed in the loop of Henle, primarily in the thick ascending limb. In the thin descending and ascending limbs of the Henle loop, sodium and Cl− are passively reabsorbed. In the thick ascending limb, sodium crosses the luminal membranes via the Na+-H+ antiporter and by an Na+-K+-2Cl− cotransporter.123 This Na+-K+-2Cl− cotransporter is the site of action of the loop diuretics furosemide and bumetanide. There is a strong electrochemical gradient for Na+ entry across the luminal membrane in this region (i.e., strongly lumen-positive transepithelial potential difference and high luminal sodium concentration).
Approximately 5% of the filtered load of sodium is reabsorbed in the distal convoluted tubule and connecting segment. In the early distal tubule (up to the connecting segment), sodium crosses the luminal membrane by means of an Na+-Cl− cotransporter. This cotransporter is inhibited by the thiazide diuretics.37
Approximately 3% of the filtered load of sodium is reabsorbed in the collecting ducts, and this segment of the nephron is responsible for altering sodium reabsorption in response to dietary fluctuations. In the late distal tubule (so-called connecting segment) and collecting ducts, sodium enters passively through Na+ channels in the luminal membranes of the principal cells.127,147 This movement of Na+ generates a lumen-negative transepithelial potential difference that facilitates Cl− reabsorption. The Na+ channel in the principal cells is blocked by the diuretics amiloride and triamterene. One of the main effects of aldosterone is to increase the number of open luminal Na+ channels in the cortical collecting ducts, thus altering sodium reabsorption in response to changes in dietary sodium intake. The renal tubular mechanisms for sodium reabsorption are summarized in Figure 3-2.
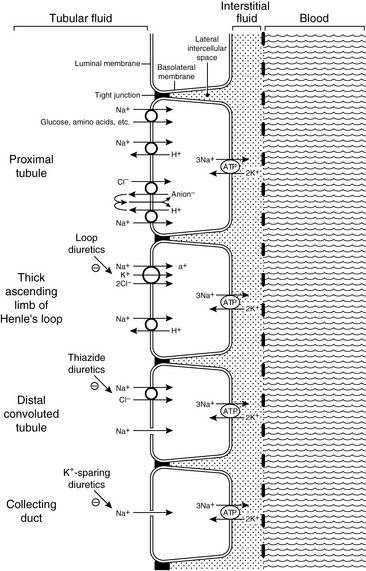
Renal regulation of sodium balance
ECF volume is directly dependent on body sodium content. The body is able to sense and respond to very small changes in sodium content. The adequacy of body sodium content is perceived as the fullness of the circulating blood volume. The term effective circulating volume has been used to refer to the relative fullness of the circulating portion of the extracellular compartment as perceived by the body. There are several sensors in the afferent limb of the body’s regulatory system for control of sodium balance (see Table 3-1). Low-pressure mechanoreceptors (i.e., volume receptors) in the cardiac atria and pulmonary vessels and high-pressure baroreceptors (i.e., pressure receptors) in the aortic arch and carotid sinus play a primary role in the body’s ability to sense the adequacy of the circulating volume. Within the kidneys, the juxtaglomerular apparatus responds to changes in perfusion pressure with changes in renin production and release. Less well characterized are receptors in the liver and the central nervous system that may contribute to sodium homeostasis.
The kidneys constitute the primary efferent limb of sodium control and regulate sodium balance by excreting an amount of sodium each day equal to that ingested. There are several overlapping control mechanisms for regulation of renal handling of sodium. This redundancy serves to protect against sodium imbalance should one control mechanism fail. The two points of control for sodium balance in the kidneys are glomerular filtration and tubular reabsorption. Autoregulation maintains renal blood flow and glomerular filtration rate (GFR) relatively constant despite fluctuations in systemic arterial pressure; thus, the filtered load of sodium is also kept relatively constant (see Chapter 2).
Glomerulotubular Balance
One mechanism is related to the fact that much of the sodium in the proximal tubules is reabsorbed along with several other solutes (e.g., glucose, amino acids, phosphate, and bicarbonate). A spontaneous increase in GFR increases the filtered load of all of these solutes, and their increased concentration in the proximal tubule enhances sodium reabsorption. Changes in peritubular capillary hydrostatic and oncotic pressures probably also play an important role in glomerulotubular balance. If GFR spontaneously increases without a change in renal plasma flow (RPF) (i.e., the filtration fraction increases), the blood leaving the efferent arterioles has lower hydrostatic pressure and higher oncotic pressure, thus favoring water and solute reabsorption in the proximal tubules (Fig. 3-3). Autoregulation (see Chapter 2) also contributes to glomerulotubular balance. When renal perfusion pressure is increased, afferent arteriolar constriction prevents transmission of the increased hydrostatic pressure to the glomerular capillaries and minimizes any increase in GFR and filtered solute load.
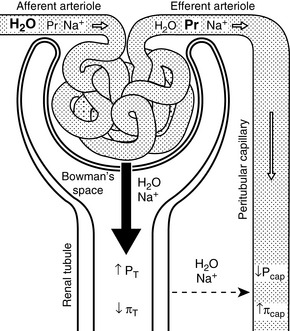
Peritubular Capillary Factors (Starling Forces)
Increased sodium intake leads to expansion of the ECF volume and compensatory increases in both GFR and RPF (i.e., the filtration fraction remains unchanged). This increases hydrostatic pressure and decreases oncotic pressure in the peritubular capillaries, thus reducing sodium and water reabsorption in the proximal tubules. Decreased sodium intake leads to volume contraction. In this setting, RPF decreases more than GFR (i.e., the filtration fraction increases). This results in decreased hydrostatic and increased oncotic pressures in the peritubular capillaries and enhanced proximal tubular reabsorption of sodium and water (see Fig. 3-3).
Angiotensin II
Decreased perfusion pressure in the afferent arterioles increases renin release from the granular cells of the juxtaglomerular apparatus and initiates the cascade of events leading to production of angiotensin II. Angiotensin II-induced vasoconstriction causes efferent more than afferent arteriolar constriction, which results in an increase in filtration fraction and changes in peritubular capillary Starling forces (decreased hydrostatic pressure and increased oncotic pressure) that facilitate proximal tubular reabsorption of sodium and water (see Fig. 3-3). Angiotensin II also directly stimulates the Na+-H+ antiporter in the proximal tubules, which facilitates sodium reabsorption and stimulates secretion of aldosterone from the adrenal gland.
Atrial Natriuretic Peptide
Atrial natriuretic peptide is one member of a family of natriuretic proteins that also includes brain natriuretic peptide (which ironically predominates in the cardiac ventricles) and C-type natriuretic peptide in the central nervous system.97 Atrial natriuretic peptide is synthesized and stored in atrial myocytes until it is released in response to atrial distention caused by volume expansion. It has a number of effects that facilitate renal excretion of sodium. Atrial natriuretic peptide causes dilation of the afferent arterioles and constriction of the efferent arterioles, leading to a primary increase in the GFR. It relaxes mesangial cells, resulting in an increase in the glomerular surface area available for filtration. Atrial natriuretic peptide also inhibits sodium reabsorption in the cortical and inner medullary collecting ducts and inhibits renin secretion, thereby decreasing production of angiotensin II and limiting the effects of angiotensin II on proximal tubular sodium reabsorption. Finally, it inhibits aldosterone secretion by adrenal zona glomerulosa.
Pressure Natriuresis
Renal sodium excretion and water excretion are markedly increased when renal arterial pressure increases even slightly without a change in the GFR. The mechanism for pressure natriuresis appears to be entirely intrarenal and does not require neural or endocrine input (i.e., it occurs in the isolated denervated kidney). The effectors of sodium balance are summarized in Table 3-3.

Regulation of water balance
The osmolality of ECF and serum sodium concentration are regulated by adjusting water balance. Osmoreceptors in the hypothalamus constitute the afferent limb (sensors) for regulation of water balance. Vasopressin (antidiuretic hormone) release is stimulated when the osmoreceptors shrink in response to plasma hyperosmolality and is inhibited when they swell in response to plasma hypoosmolality. Vasopressin (water output) and thirst (water input) constitute the efferent limb (effectors) for the regulation of water balance (see Table 3-1).
Vasopressin (Antidiuretic Hormone)
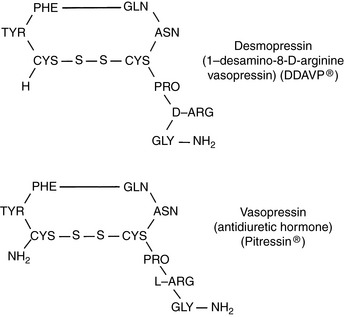
Vasopressin (antidiuretic hormone [ADH]) is a nine-amino acid peptide synthesized in neurons of the supraoptic and paraventricular nuclei in the hypothalamus (Fig. 3-4). It travels down the axons of these neurons and is released into the circulation at the level of the neurohypophysis.
Vasopressin increases the reabsorption of water in the collecting ducts of the kidneys and increases the permeability of the medullary collecting ducts to urea.1 Vasopressin attaches to V2 receptors on the basolateral membranes of the principal cells of the cortical and medullary collecting ducts. The hormone-receptor complex activates a guanine nucleotide regulatory protein (Gs), resulting in replacement of guanosine diphosphate (GDP) with guanosine triphosphate (GTP) and stimulation of adenyl cyclase in the cell membrane. Formation of cyclic adenosine monophosphate (cAMP) results in activation of protein kinase A, which in turn phosphorylates a specific serine residue on subunits of the tetrameric aquaporin 2 (AQP2) proteins found in membranes of subapical vesicles in the cytoplasm of the principal cells. Phosphorylation results in trafficking and insertion of AQP2 water channels into the luminal membranes of the principal cells.121,164 When vasopressin is absent or in low concentration, AQP2 channels are removed from the luminal membrane by endocytosis. Aquaporin 3 (AQP3) and 4 (AQP4) channels are found in the basolateral membranes of the principals cells and represent exit pathways for water that enters the cells via the luminal AQP2 channels. The AQP3 channel is found in the cortical and outer medullary collecting ducts, whereas AQP4 is located primarily in the inner medullary collecting ducts. In the absence of vasopressin, urine osmolality can be decreased to as low as 50 mOsm/kg by continued reabsorption of sodium without water as tubular fluid passes down the collecting ducts. The V1A receptors are located in vascular smooth muscle and cause vasoconstriction when AVP binds to them. V1B receptors are found primarily in the hypothalamus where AVP binding leads to increased secretion of corticotropin.
The effect of vasopressin on urea reabsorption may be important in the pathogenesis of medullary washout of solute in chronic polyuric states. Chronic diuresis can lead to depletion of urea from the medullary interstitium by suppression of vasopressin release and impaired urea reabsorption in the medullary collecting ducts. During antidiuresis, urea may constitute more than 40% of the medullary solute. During diuresis, however, it may constitute less than 10% of the medullary solute.17,98 The urinary concentrating mechanism is discussed in Chapter 2.
Stimuli for Vasopressin Release
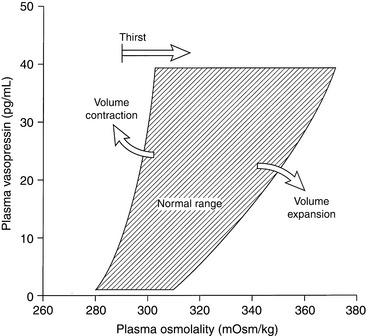
The major stimulus for vasopressin release is hypertonicity of plasma reaching the osmoreceptors of the hypothalamus. The threshold for vasopressin release in humans corresponds to a plasma osmolality of 280 mOsm/kg, and similar or slightly higher threshold values have been observed in healthy experimental dogs.31,135,137 Below this osmolality, vasopressin release is suppressed, and urine is maximally diluted. One hour after oral administration of water at 40 mL/kg, normal dogs developed a mean urine osmolality of 132 mOsm/kg (range, 68 to 244 mOsm/kg).67 In humans, the release of vasopressin is maximal at a plasma osmolality of 294 mOsm/kg, and at this plasma osmolality the thirst mechanism becomes operative.137 Thus, changes in plasma osmolality as small as 1% to 2% above normal lead to maximal vasopressin release. The gain of the system is such that a 1 mOsm/kg increase in plasma osmolality leads to an almost 100 mOsm/kg increase in urine osmolality. The vasopressin system curtails water excretion, but further defense against hypertonicity requires a normal thirst mechanism and access to water. The thirst mechanism has both osmoreceptors and volume receptors. The volume receptors for the thirst mechanism are stimulated by angiotensin II and may be under control of the renin-angiotensin system.108
The next most important stimulus for vasopressin release is volume depletion sensed by baroreceptors in the left atrium, aortic sinus, and carotid sinuses. A decrease in blood volume of 5% to 10% lowers the threshold for vasopressin release and increases the sensitivity of the osmoregulatory mechanism (Fig. 3-5).59,137 Nonosmotic stimulation of vasopressin by actual or perceived volume depletion plays a major role in the generation and perpetuation of hyponatremia in states of true volume depletion and in some conditions (e.g., heart failure, liver failure, nephrotic syndrome) associated with hypervolemia (see Hypovolemic Hyponatremia and Hypervolemic Hyponatremia sections).
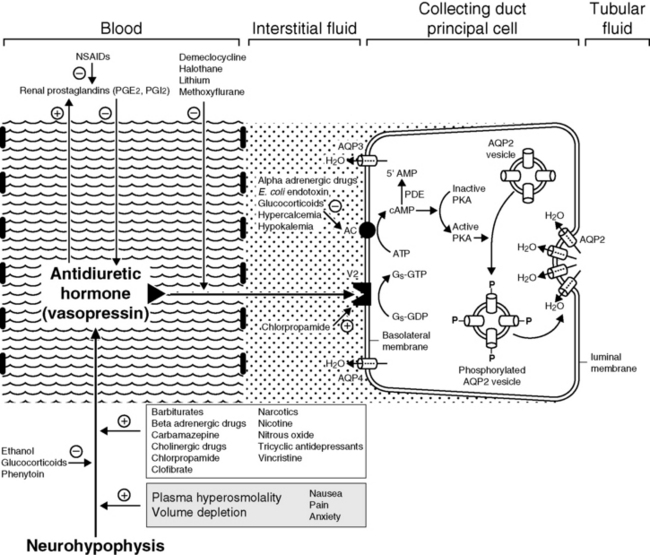
Other stimuli for vasopressin release include nausea, pain, and emotional anxiety. Many drugs and some electrolyte disturbances affect the release and renal action of vasopressin. The effects of some of these are depicted in Figure 3-6.
Role of the Kidneys in Water Balance
Three conditions must be met for the kidneys to excrete a water load normally. First, there must be adequate delivery of tubular fluid to distal diluting sites (ascending limb of Henle’s loop) where NaCl is removed without water, rendering the tubular fluid hypotonic to the medullary interstitium. Adequate distal delivery requires a normal RPF, normal GFR, and normal isosmotic reabsorption of sodium and water from the proximal tubules mediated by aquaporin 1 (AQP1) channels in the luminal and basolateral membranes of these cells. In the presence of volume depletion, RPF is usually decreased more than the GFR, and enhanced proximal tubular reabsorption of sodium and water may result from changes in postglomerular hemodynamics (see Fig. 3-3). These factors may prevent adequate distal delivery of tubular fluid for dilution.
In the absence of vasopressin, the collecting ducts remain impermeable to water, the urine becomes maximally dilute, and polyuria develops. Hypertonicity and hypernatremia occur if the animal is unable to drink enough water to balance the tremendous loss of water in the urine. Hypertonicity and hypernatremia also may develop in states of osmotic diuresis (e.g., diabetes mellitus, mannitol administration, chronic renal failure, postobstructive diuresis). Urine osmolality approaches plasma osmolality during osmotic diuresis, and the solute responsible for the diuresis displaces sodium and other electrolytes in urine.51 Hypertonicity develops to the extent that displaced sodium remains in the ECF.
Defense Against Hypotonicity
It is crucial to the survival of the animal that the brain be protected against changes in plasma tonicity, because an increase in brain water content of more than 10% is incompatible with life.151 The fact that animals with chronic hyponatremia may have serum sodium concentrations that are 10% or more below normal attests to the brain’s ability to adapt to hypotonicity. For example, based on osmotic considerations alone, a decrease in serum sodium concentration from 145 to 132 mEq/L would correspond to an increase in intracellular water of 10%. During acute hypotonicity, water moves into the brain. The increase in hydrostatic pressure in the interstitial compartment of the brain immediately forces sodium-containing ECF into the cerebrospinal fluid. This movement of fluid out of the brain occurs within minutes and limits the change in brain water content to much less than would be anticipated based on osmotic considerations alone.151 During the first 24 hours of hypotonicity, movement of potassium out of cells also contributes substantially to the protection of the brain from an acute decrease in plasma osmolality. After 24 to 48 hours, a reduction in the cellular content of organic solutes contributes to the brain’s defense against hypotonicity. These organic osmolytes are substances that can be used by cells to maintain intracellular tonicity without having adverse effects on cellular metabolism and include amino acids (e.g., taurine, glutamate, and glutamine), methylamines (e.g., phosphocreatine), and polyols (e.g., myoinositol).6,134 The very devices that protect the brain against plasma hypotonicity predispose it to injury when hyponatremia is corrected. Solutes lost during adaptation must be recovered, and this process requires several days. If correction of hyponatremia proceeds more quickly than recovery of lost solutes can occur, a devastating complication of treatment called osmotic demyelination syndrome (myelinolysis) may occur (see Treatment of Hyponatremia section).
Clinical approach to the patient with hypernatremia
Hypernatremia is less common than hyponatremia. Intense thirst normally protects against development of hypernatremia unless water is not available or a neurologic disorder is present that either prevents access to water or interferes with recognition of thirst. All clinical conditions associated with hypernatremia reflect hyperosmolality and hypertonicity of the ECF if the solute in question is impermeant. A deficit of pure water, loss of hypotonic fluids, or gain of sodium can cause hypertonicity of the ECF and hypernatremia. The causes of hypernatremia are listed in Box 3-1, and the clinical approach to the patient with hypernatremia is outlined in Figure 3-7.
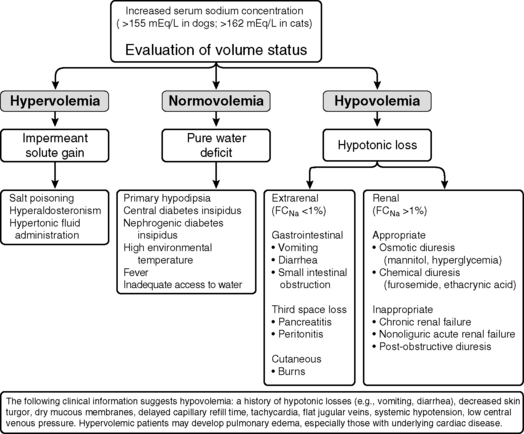
Figure 3-7 Clinical approach to the patient with hypernatremia. FCNa, Fractional clearance of sodium.
Pure water loss
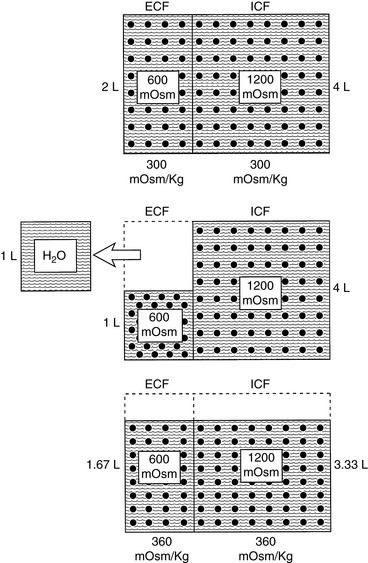
When a deficit of pure water develops, the ECF becomes hypertonic in relation to the intracellular fluid (ICF), and osmotic forces cause movement of water from the intracellular to the extracellular compartment. The result is that the volume loss is shared proportionately between the extracellular and intracellular compartments. Approximately two thirds of the volume loss comes from the intracellular compartment and one third from the extracellular compartment. Plasma volume is one fourth of the ECF, and thus one twelfth of the volume loss (¼ × ⅓) is derived from the intravascular space. The oncotic pressure generated by plasma proteins favors retention of water within vessels, and the plasma compartment may not share proportionately in the volume loss.41 As a result of these factors, volume depletion is usually not a clinical feature of pure water loss. It is almost impossible for a conscious animal with an intact thirst mechanism and access to water to develop hypertonicity caused by pure water loss. Thus, hypertonicity associated with pure water loss usually implies that water intake has been defective.
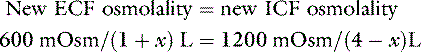
where x is the volume of water moving between compartments:
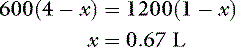
The new volumes and osmolalities are:
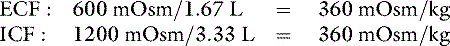
Note that the intracellular compartment has lost an amount equal to two thirds of the water deficit (0.67 L) and that the final ECF volume (1.67 L) is lower than the original volume (2 L) by an amount equal to one third of the total water deficit (0.33 L). Thus, the two compartments have shared proportionately in the water loss. These changes are depicted in Figure 3-8.
Rarely, chronic hypernatremia may occur in fully conscious animals that have access to water. In these cases, abnormal osmoregulation of ADH release caused by underlying hypothalamic lesions results in hypodipsia. Animals that are unable to obtain water because central nervous system disease has resulted in an altered sensorium may also be hypernatremic; but in these instances, the hypernatremia is simply a result of water deprivation. Hypodipsic hypernatremia related to defective osmoregulation of ADH has been reported in a dog with hydrocephalus and normal pituitary function.31 In normal individuals, administration of hypertonic saline increases plasma osmolality and simultaneously causes volume expansion. Osmoreceptors are stimulated by hyperosmolality but inhibited by volume expansion. Normally, the response to hyperosmolality takes precedence, and ADH secretion increases, resulting in decreased urine volume and increased urine osmolality. The affected dog experienced increased urine volume and decreased urine osmolality in response to an infusion of hypertonic saline, indicating defective osmoreceptor function as observed in human patients with hypodipsic hypernatremia. Similarly, destruction of osmoreceptors in the hypothalamus was thought to be responsible for adipsia and hypernatremia in a dog with focal granulomatous meningoencephalitis.104 Weakness and polymyopathy have been reported in a young cat with hypodipsia, hypernatremia, and hypertonicity associated with hydrocephalus and hypopituitarism, and hypernatremia, adipsia, and diabetes insipidus have been observed in a young dalmatian dog with dysplasia of the rostral diencephalon.5,34 Hypernatremia also has been reported in a dog63 and cat115 with central nervous system lymphoma.
Hypodipsia, hypernatremia, and hypertonicity caused by an abnormal thirst mechanism have been reported in young female miniature schnauzers and in a young Great Dane.27,70,76,112,159 One miniature schnauzer with hypodipsic hypernatremia had severe behavioral disturbances, and holoprosencephaly was found at necropsy.153 Another had dysgenesis of the corpus callosum and other forebrain structures.112 Grossly visible neuroanatomic abnormalities were not identified in a previous report.27 Whether a spectrum of neuroanatomic abnormalities exists in these dogs (which appear to have a form of congenital adipsic hypernatremia) is not known. Infusion of hypertonic saline has been shown to lead to an increase in urine volume and a decrease in urine osmolality compatible with defective osmoregulation of ADH.27 Clinical signs in affected dogs are associated with hypertonicity and include anorexia, lethargy, weakness, disorientation, ataxia, and seizures. Affected dogs can be managed clinically by addition of water to their food, but hypernatremia and neurologic dysfunction recur whenever water supplementation is discontinued. In a Norwegian elkhound with adipsic hypernatremia, the adipsia resolved spontaneously at 2 years of age,62 and an adipsic Labrador retriever with hypothyroidism responded to treatment with levothyroxine.81
Central or pituitary diabetes insipidus (CDI) is caused by a partial or complete lack of vasopressin production and release from the neurohypophysis.65 It may result from trauma or neoplasia or may be idiopathic in dogs and cats.⁎ Visceral larva migrans also has been reported to cause CDI in a dog.99 In one dog with hypernatremia, hypertonicity, and gastric dilation-volvulus, CDI was present and caused by neurohypophyseal atrophy secondary to a cystic craniopharyngeal duct.36 Congenital CDI is rare58,86,163 but has been reported in two sibling Afghan pups.131 Traumatic CDI may be transient in nature. Hypophysectomy for treatment of hyperadrenocorticism results in transient CDI that may take several weeks to resolve.102 Marked hypernatremia occurs in dogs in the first 24 hours after hypophysectomy and can be prevented by prophylactic treatment with desmopressin (DDAVP).64 In the month after surgery, serum sodium concentrations in control dogs were not markedly different from those observed in the DDAVP-treated group, suggesting that the dogs with untreated CDI drank sufficient water to maintain relatively normal plasma osmolality. The transient nature of CDI after hypophysectomy may result from the fact that some of the vasopressin-producing neurons from the hypothalamus terminate in the median eminence.
Animals with CDI have severe polydipsia and polyuria. Their urine typically is hyposthenuric (urine osmolality, 60 to 200 mOsm/kg), but urine osmolality may approach 400 to 500 mOsm/kg in the presence of dehydration. Variability in USG and urine osmolality values at the time of presentation in dogs and cats with diabetes insipidus presumably is related to hydration status and severity of vasopressin deficiency. In one study, dogs were classified as having complete or partial CDI based on the magnitude of increase in their USG and urine osmolality after induction of 5% dehydration.65 Dogs with complete CDI had USG values of 1.001 to 1.007 that did not change substantially after induction of 5% dehydration, whereas dogs with partial CDI had USG values of 1.002 to 1.016 that increased to 1.010 to 1.018 after induction of 5% dehydration. In both groups, there was a substantial (>50%) increase in USG 2 hours after administration of 1 to 5 U of aqueous arginine vasopressin. Affected dogs responded well to administration of DDAVP acetate (1 to 2 drops in both eyes every 12 to 24 hours), but the prognosis was dependent on the underlying cause of CDI. Many older dogs with CDI had tumors in the region of the pituitary gland and developed neurologic signs.
Increased plasma osmolality and hypernatremia may occur in dogs and cats with CDI. These results suggest that some affected dogs and cats do not obtain enough water to maintain water balance and are presented in a hypertonic state. Severe hypernatremia and neurologic dysfunction may occur if the animal cannot maintain adequate water intake.36,133 In contrast, with psychogenic polydipsia, plasma osmolality and serum sodium concentration may be lower than normal at presentation.91 Administration of vasopressin leads to an increase in urine osmolality or specific gravity in dogs and cats with CDI, but the initial response may be less than expected because of renal medullary washout of solute. In one study, USG values increased to 1.018 to 1.022 after vasopressin administration in dogs with complete CDI and to 1.018 to 1.036 in dogs with partial CDI.65
DDAVP is a structural analogue of vasopressin (see Fig. 3-4) that has a more potent antidiuretic effect than vasopressin but a minimal vasopressive effect and is relatively resistant to metabolic degradation. DDAVP is available as a nasal spray (0.1 mg/mL), injectable solution (4 µg/mL), or tablet for oral administration (0.1 and 0.2 mg). The injectable solution is much more expensive than the nasal spray, and the nasal spray has been used subcutaneously in dogs and in a cat with CDI at a dosage of 1 µg/kg without adverse effects.86,87 Polyuria and polydipsia in a cat with CDI were controlled with 1 µg/kg administered subcutaneously every 12 hours or 1.5 µg/kg administered conjunctivally every 8 hours. One drop of the nasal spray contains 1.5 to 4 µg of DDAVP, and the duration of effect varies from 8 to 24 hours.43 In humans, the bioavailability of DDAVP after oral administration was 0.1% as compared with 3% to 5% after intranasal administration, and gastrointestinal absorption was improved when it was given in a fasted state.46,136 In dogs, an antidiuretic effect was observed even after orally administered doses as low as 50 µg.160
Chlorpropamide is a sulfonylurea hypoglycemic agent that potentiates the renal tubular effects of small amounts of vasopressin and may be useful in management of animals with partial CDI. Its effect may occur by up-regulation of ADH receptors in the kidneys.35 The recommended dosage of chlorpropamide is 10 to 40 mg/kg/day orally, and hypoglycemia is a potential adverse effect. It has been useful in the management of CDI (up to 50% reduction in urine output) in some reports but not in others, possibly because some animals have partial and some have complete CDI.86,140
In the broadest sense, the term nephrogenic diabetes insipidus (NDI) may be used to describe a diverse group of disorders in which structural or functional abnormalities interfere with the ability of the kidneys to concentrate urine (Box 3-2).13,90 Congenital NDI is a rare disorder in small animal medicine.13,80,90 Affected animals are presented at a very young age for severe polyuria and polydipsia. In reported cases, urine osmolality and specific gravity have been in the hyposthenuric range. Affected animals show no response to water deprivation testing, exogenous vasopressin administration, or hypertonic saline infusion. In one case report, the plasma vasopressin concentration was markedly increased.80 Congential NDI in human patients can arise from mutations in the V2 receptor (X-linked recessive inheritance) or from mutations in the AQP2 channel (autosomal recessive inheritance). Low affinity V2 receptors were thought to be responsible for congenital NDI in a family of Siberian huskies.103



