Chapter 5 Disorders of Potassium
Hypokalemia and Hyperkalemia
Potassium is the major intracellular cation in mammalian cells, whereas sodium is the major extracellular cation. Normally, the extracellular fluid (ECF) sodium concentration is approximately 140 mEq/L, and the ECF potassium concentration is approximately 4 mEq/L. This relationship is reversed in intracellular fluid (ICF), in which the sodium concentration is approximately 10 mEq/L and the potassium concentration is approximately 140 mEq/L. In experimental studies of dogs, control values for ICF sodium and potassium concentrations in skeletal muscle were 8.4 to 13.7 and 139 to 142 mEq/L, respectively.20,107
Total body potassium content in humans is approximately 50 to 55 mEq/kg body weight, and almost all of this potassium is readily exchangeable.6,70 In one study of potassium depletion in dogs, the control value for total exchangeable potassium as determined by 50K dilution was 47.1 mEq/kg body weight (range, 39.8 to 61.1 mEq/kg).1 In cats, total body potassium is approximately 55 mEg/kg body weight.184a As much as 95% or more of total body potassium is located within cells, with muscle containing 60% to 75% of this potassium. Muscle potassium content in normal dogs and cats is approximately 400 mEq/kg.20,107,147,191 As a solute, intracellular potassium is crucial for maintenance of normal cell volume. Intracellular potassium also is important for normal cell growth because it is required for the normal function of enzymes responsible for nucleic acid, glycogen, and protein synthesis.
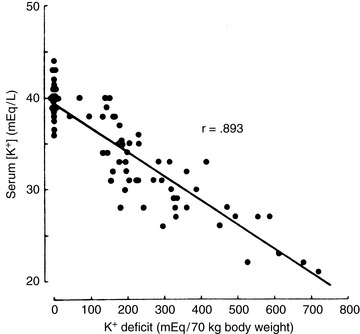
The remaining 5% of the body’s potassium is located in the ECF. Maintaining the ECF potassium concentration within narrow limits is critical to avoid the life-threatening effects of hyperkalemia on cardiac conduction. In humans, the serum potassium concentration is inversely correlated with the total body deficit of potassium (Fig. 5-1). Likewise, in dogs with potassium depletion induced by dietary restriction, the muscle potassium content was strongly correlated (r = 0.87) with the serum potassium concentration.147 During translocation of potassium between ICF and ECF, however, the serum potassium concentration can change without any change in the total body potassium content. One of the most important functions of potassium in the body is its role in generation of the normal resting cell membrane potential.
The resting cell membrane potential
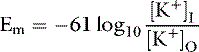

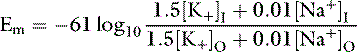
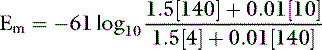
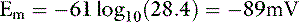
In one study of dogs with potassium deficiency, the predicted Em was −86.6 mV and the measured Em in skeletal muscle of control animals was −90.1 mV.20 The resting cell membrane potential plays a vital role in the normal function of skeletal and cardiac muscle, nerves, and transporting epithelia.
The threshold cell membrane potential
Hypokalemia increases the resting potential (i.e., makes it more negative) and hyperpolarizes the cell, whereas hyperkalemia decreases the resting potential (i.e., makes it less negative) and initially makes the cell hyperexcitable (Fig. 5-2). If the resting potential decreases to less than the threshold potential, depolarization results, repolarization cannot occur, and the cell is no longer excitable. Translocation of potassium between body compartments results in a greater change in the ratio of intracellular to extracellular potassium concentrations ([K+]I/[K+]O) than does a change in total body potassium. In the former instance, the potassium concentrations of the two compartments change in opposite directions, whereas in the latter instance, they change in the same direction.

Membrane excitability also is affected by ionized calcium concentration and acid-base balance. Calcium affects the threshold potential rather than the resting potential. Ionized hypocalcemia increases membrane excitability by allowing self-perpetuating sodium permeability to be reached with a lesser degree of depolarization, whereas ionized hypercalcemia requires greater than normal depolarization for this threshold to be reached (see Fig. 5-2). Thus, hypercalcemia counteracts hyperkalemia by normalizing the difference between the resting and threshold potentials, whereas hypocalcemia exacerbates the effect of hyperkalemia on membrane excitability. This principle is the basis for treating hyperkalemia with calcium salts (see the Treatment of Hyperkalemia section). Membrane excitability is increased by alkalemia and decreased by acidemia. As a result of these factors, clinical signs are not necessarily correlated with serum potassium concentrations. Electrocardiographic findings and muscle strength reflect the functional consequences of abnormalities in serum potassium concentration.
Potassium balance
External potassium balance
External balance for potassium is maintained by matching output (primarily in urine) to input (from the diet). In the normal animal, potassium enters the body only through the gastrointestinal tract, and virtually all ingested potassium is absorbed in the stomach and small intestine. Transport of potassium in the small intestine is passive, whereas active transport (responsive to aldosterone) occurs in the colon. Colonic secretion of potassium may play an important role in extrarenal potassium homeostasis in some disease states (e.g., chronic renal failure) (Fig. 5-3).
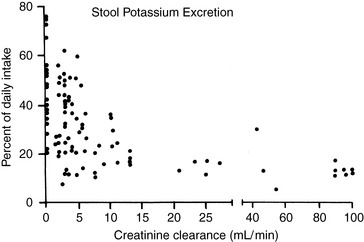
Potassium derived from the diet and endogenous cellular breakdown is removed from the body primarily by the kidneys and, to a much lesser extent, by the gastrointestinal tract. During zero balance, 90% to 95% of ingested potassium is excreted in urine, and the remaining 5% to 10% is excreted via the gastrointestinal tract. This pattern of output has been observed during control balance studies in normal dogs.12,139,152,169,182 In a study of renal handling of potassium in dogs, 90% to 98% of potassium intake was eliminated from the body by the kidneys.24
Adaptation occurs during chronic potassium loading so that the animal is protected from hyperkalemia that could occur as a result of an acute potassium load. This effect results from enhanced renal and colonic excretion of potassium, as well as from enhanced uptake of potassium by the liver and muscle, mediated by the effects of insulin and catecholamines. Potassium deprivation is associated with decreased aldosterone secretion, suppression of potassium secretion in the distal nephron, and increased reabsorption of potassium in the inner medullary collecting ducts. Skeletal muscle potassium concentration decreases, but brain and heart potassium concentrations are minimally affected during potassium depletion.20,107,178 The colon adapts to potassium deprivation by decreasing its secretion of potassium.
Internal potassium balance
Internal balance for potassium is maintained by translocation of potassium between ECF and ICF. One half to two thirds of an acute potassium load appears in the urine within the first 4 to 6 hours, and effective translocation of potassium from ECF to ICF is crucial in preventing life-threatening hyperkalemia until the kidneys have sufficient time to excrete the remainder of the potassium load. Endogenous insulin secretion and stimulation of β2-adrenergic receptors by epinephrine promote cellular uptake of potassium in the liver and muscle by increasing the activity of cell membrane Na+, K+-ATPase. The main effects of these hormones are to facilitate distribution of an acute potassium load and not to mediate minor adjustments in serum potassium concentration. The ECF concentration of potassium itself plays an important role in translocation because potassium movement into cells is facilitated by the change in chemical concentration gradient resulting from addition of potassium to ECF. The fraction of an acute potassium load taken up by the body is increased during chronic potassium depletion and decreased when total body potassium is excessive. In summary, any change in serum potassium concentration must arise from a change in intake, distribution, or excretion (Fig. 5-4).
Effect of acid-base balance on potassium distribution
The effect of acute pH changes on translocation of potassium between ICF and ECF is complex. In general, acidosis is associated with movement of potassium ions from ICF to ECF, and alkalosis is associated with movement of potassium ions from ECF to ICF. Early animal studies and observations in a small number of human patients led to the prediction that acute metabolic acidosis would be associated with a 0.6-mEq/L increment in serum potassium concentration for each 0.1-U decrement in pH. This rule of thumb has circulated widely among clinicians.31,181,188
However, a critical review of experimental studies in animals and humans demonstrated that changes in serum potassium concentration during acute acid-base disturbances were quite variable.4 The change in serum potassium concentration was greatest during acute mineral acidosis. In dogs, the increase in serum potassium concentration after administration of a mineral acid (e.g., HCl or NH4Cl) was very variable, ranging from a 0.17- to 1.67-mEq/L increment in serum potassium concentration per 0.1-U decrement in pH (mean, 0.75 mEq/L). The increment in serum potassium concentration during acute respiratory acidosis in dogs was much lower, averaging only 0.14 mEq/L per 0.1-U decrement in pH. The decrement in serum potassium concentration during metabolic alkalosis in dogs averaged 0.18 mEq/L per 0.1-U increment in pH, whereas it averaged 0.27 mEq/L per 0.1-U increment in pH during respiratory alkalosis. In another study, respiratory alkalosis induced by hyperventilation in anesthetized dogs caused a somewhat greater decrement in serum potassium concentration (0.4 mEq/L) for each 0.1-U increment in pH.136 An increase in serum potassium concentration did not occur in acute metabolic acidosis caused by organic acids (e.g., lactic acid and ketoacids).* Acute infusion of β-hydroxybutyric acid in normal dogs caused an increase in insulin in portal venous blood and hypokalemia, presumably as a result of potassium uptake by cells. Conversely, acute infusion of HCl led to increased portal vein glucagon concentration and hyperkalemia, possibly caused by potassium release from cells.5 In summary, only mineral acidosis is expected to cause any clinically relevant change in serum potassium concentration during acute acid-base disturbances.
Many factors probably contribute to the variable changes observed in serum potassium concentration during acute acid-base disturbances, including blood pH and HCO3− concentration, nature of the acid anion (mineral versus organic), osmolality, hormonal activity (e.g., catecholamines, insulin, glucagon, and aldosterone), and the metabolic and excretory roles of the liver and kidneys.4 Hyperosmolality and lack of insulin are more likely to be responsible for hyperkalemia observed in patients with diabetic ketoacidosis than is the acidosis itself.
Hyperkalemia associated with acute metabolic acidosis induced by mineral acids is transient. In a study of acute and chronic metabolic acidosis induced in dogs by administration of HCl or NH4Cl, hyperkalemia was observed after acute infusion of HCl, but hypokalemia developed after 3 to 5 days of NH4Cl administration.122 The observed hypokalemia was associated with inappropriately high urinary excretion of potassium and increased plasma aldosterone concentration.122 Similar findings have been reported in rats with chronic metabolic acidosis induced by NH4Cl. Despite a total body deficit of potassium, rats with chronic metabolic acidosis did not conserve potassium appropriately.170 This effect may be caused by a decreased filtered load of HCO3−, increased distal delivery of sodium, and increased distal tubular flow. Thus, metabolic acidosis of at least 2 to 3 days’ duration is associated with increased urinary potassium excretion and mild hypokalemia rather than hyperkalemia.79
Renal handling of potassium
Mechanisms of renal tubular transport of potassium
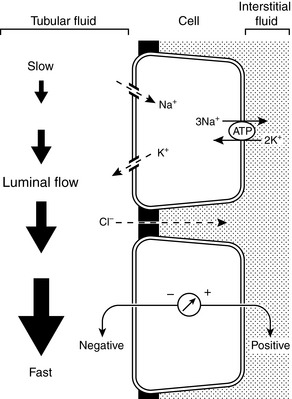
The transepithelial electrical potential difference is lumen negative in the early proximal tubule, but no active transport mechanism for potassium has been discovered in this segment of the nephron. In the proximal tubule, potassium is reabsorbed along with water by solvent drag via the paracellular route. Apparently, water reabsorption increases the luminal concentration of potassium enough to overcome the unfavorable transepithelial potential difference. The transepithelial electrical potential difference becomes lumen positive in the late proximal tubule, and this facilitates reabsorption of potassium by the paracellular route. Transcellular transport of potassium in the proximal tubular cells occurs by means of potassium channels in both luminal and basolateral membranes and by a K+-Cl− cotransporter in basolateral membranes (Fig. 5-5).
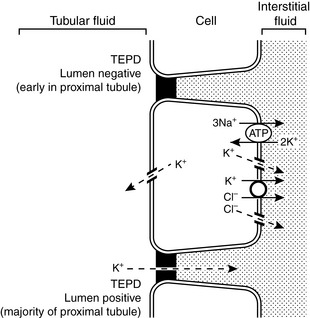
Figure 5-5 Renal tubular transport mechanisms for potassium in the proximal tubule.
TEPD, Transepithelial potential difference. (Drawing by Tim Vojt.)
In the thick ascending limb of the Henle loop, the transepithelial electrical potential difference is strongly lumen positive, and most potassium reabsorption occurs by the paracellular route. Potassium channels in the luminal membranes allow potassium to exit the cell down its concentration gradient and facilitate the electrochemical gradient for potassium reabsorption via the paracellular route. Transcellular reabsorption of potassium is facilitated by the luminal Na+-K+-2Cl− cotransporter and by potassium channels and a K+-Cl− cotransporter in the basolateral membranes (Fig. 5-6).
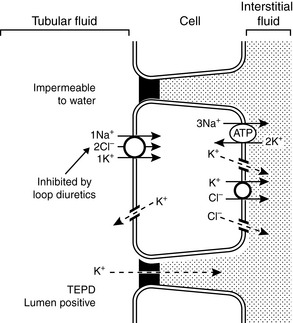
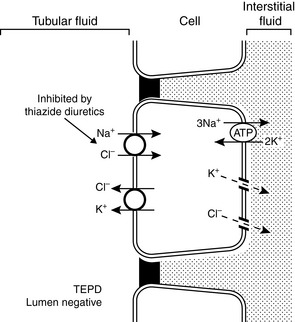
The mechanisms of renal potassium handling in the distal convoluted tubule are shown in Figure 5-7. The thiazide-sensitive Na+-Cl− cotransporter and the K+-Cl− cotransporter in the luminal membranes of these tubular cells result in secretion of potassium and reabsorption of sodium while chloride is recycled across the luminal membrane. The basolateral Na+, K+-ATPase maintains a low intracellular concentration of sodium and a high intracellular concentration of potassium that facilitate sodium reabsorption and potassium secretion across the luminal membranes.
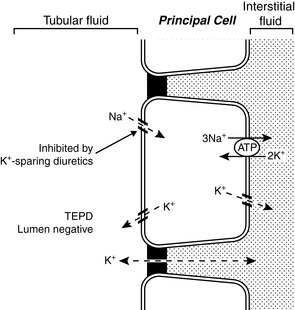
Principal cells are found in the connecting tubule and collecting duct and are responsible for potassium secretion. The basolateral membranes of principal cells are rich in Na+, K+-ATPase, which maintains a high intracellular potassium concentration. The luminal membranes of the principal cells contain an electrogenic sodium channel (ENaC). This sodium channel is directly blocked by the diuretics amiloride and triamterene, whereas spironolactone antagonizes the effect of aldosterone on the channel. Sodium movement through this channel renders the tubular lumen negative, and the resultant increase in lumen electronegativity facilitates secretion of K+ ions through luminal K+ channels (Fig. 5-8).
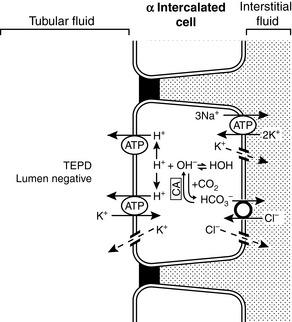
There are two types of intercalated cells in the distal nephron. Type A or α intercalated cells contain H+-ATPase and H+, K+-ATPase in their luminal membranes and Cl−– HCO3− countertransporters and Cl− and K+ channels in their basolateral membranes. They also contain carbonic anhydrase. This arrangement allows the intercalated cell to secrete H+ ions and reabsorb K+ and HCO3− ions. Potassium is actively transported across the luminal membranes of type α intercalated cells by H+, K+-ATPase and then diffuses down its concentration gradient through potassium channels in the basolateral membranes (Fig. 5-9). Type A and α intercalated cells are found in the connecting tubule, cortical collecting duct, and outer medullary collecting duct. Type B and β intercalated cells are found only in the cortical collecting ducts and secrete HCO3− ions. They are able to do so because their polarity is reversed as compared with type α intercalated cells (i.e., the H+-ATPase is in the basolateral membrane, and the Cl−– HCO3− countertransporter is in the luminal membrane).
Determinants of urinary potassium excretion
The Na+ and Cl− concentrations of distal tubular fluid usually have little effect on K+ secretion. When the luminal Na+ concentration is very low (<25 to 35 mEq/L), however, diffusion of Na+ ions into distal tubular cells may be impaired sufficiently to produce an increase in the tubular cell transmembrane potential (making the cell interior more negative) and impeding diffusion of K+ ions from the cell into the tubular lumen.85,204,205 Extremely low luminal Cl− concentrations (<10 mEq/L) may increase net potassium secretion, possibly because some fraction of K+ reabsorption or secretion may be accomplished by K+-Cl− cotransport.198 Such a mechanism may also play a role in the pathophysiology of enhanced urinary K+ excretion during metabolic alkalosis. Antidiuretic hormone (ADH) helps minimize disruption of potassium balance during water deprivation by increasing the number of open luminal K+ channels in principal cells and facilitating potassium excretion at a time when distal tubular flow is reduced.36,69,183 Conversely, potassium excretion is not necessarily increased despite increased distal tubular flow during water diuresis because ADH is suppressed. Major factors affecting renal excretion of potassium are summarized in Figure 5-10.
Factors influencing renal potassium excretion
Potassium Intake
Dietary potassium intake also has a direct effect on the function of the luminal potassium channels in principal cells. A high potassium intake increases the activity of these channels by decreasing the phosphorylation of a specific tyrosine residue in the ROMK protein component of the channel, which in turn results in decreased removal of the channels from the luminal membranes. A low potassium diet has the opposite effect.82,201,203
Hydrogen Ion Balance
Acute mineral metabolic acidosis decreases urinary excretion of potassium. Chronic metabolic acidosis actually may increase urinary excretion of potassium. If distal tubular flow remains constant, acute (<8 hours) mineral metabolic acidosis results in decreased urinary excretion of potassium because, during metabolic acidosis caused by administration of a mineral acid, H+ ions enter cells to be buffered by intracellular proteins in exchange for K+ ions that leave cells and enter the ECF.181,188 When this ion exchange occurs across the basolateral membranes of the cells of the connecting tubule and cortical collecting ducts, the resulting decreased intracellular concentration of potassium is associated with less tubular secretion of potassium because of a less favorable chemical concentration gradient.
A critical factor determining whether acute metabolic acidosis causes this exchange of H+ and K+ ions across the cell membranes is the permeability of the anion associated with the acid. Chloride ions are relatively impermeable and cannot follow the H+ ions into the cell, whereas lactate and ketoacid anions are more permeable and can follow H+ ions into the cell so that K+ ions do not need to be exchanged with H+ ions for electroneutrality. As a result, acute mineral metabolic acidosis may be associated with H+– K+ exchange across cell membranes, but acute organic metabolic acidosis is not. Chronic (>3 days) metabolic acidosis caused by administration of a mineral acid leads to mild hypokalemia, possibly caused by stimulation of aldosterone secretion by the acidosis.79,122,170 Even in acute acidosis, a decreased filtered load of bicarbonate can reduce sodium reabsorption in the proximal tubules and increase delivery of sodium and water to the distal nephron. This increases the distal tubular flow rate and enhances urinary potassium excretion.
Normal serum concentrations
Ion-selective potentiometry and flame photometry are methods used by clinical laboratories to measure sodium and potassium concentrations in body fluids. Electrolytes in plasma are excluded from the fraction of plasma (normally about 7%) that is occupied by solids (e.g., lipids and proteins) and are confined to the aqueous phase of plasma (about 93% of total plasma volume). Flame photometry and indirect potentiometry are affected by the exclusion of electrolytes from the fraction of plasma that is occupied by solids, whereas direct potentiometry is not.194 The resulting error is usually small, but for serum sodium concentration it may be clinically relevant in patients with hyperlipemia (see Chapter 3). Potassium is present in ECF at a much lower concentration than sodium, and the effect of hyperlipemia on the measured serum potassium concentration is much less apparent.
Normal values for serum potassium concentration in dogs and cats vary slightly among laboratories but are expected to be 3.5 to 5.5 mEq/L, with an average value of approximately 4.5 mEq/L. Serum potassium concentrations exceed plasma concentrations because potassium is released from platelets during the clotting process. There is a positive correlation between platelet count and serum potassium concentration in dogs.49,155 The difference between serum and plasma potassium concentrations is most pronounced in animals with thrombocytosis.50,124,155 In one study, serum potassium concentration was greater than plasma potassium concentration by a mean of 0.63 mEq/L in dogs with normal platelet counts and by a mean of 1.55 mEq/L in dogs with thrombocytosis.155
The potassium content of erythrocytes varies in mammalian species, and hemolysis can result in hyperkalemia in species that have high red cell potassium concentrations (Table 5-1). Normal adult canine and feline red cells usually contain potassium in concentrations similar to those of plasma, and hemolysis is not associated with hyperkalemia.42,48,65,93,153 In one study, storage of canine red cells in citrate-phosphate-dextrose-adenine for 40 days resulted in an increase in plasma potassium concentration from 5 to almost 9 mEq/L despite the fact that the original intracellular potassium concentration in the red cells was only 3.8 mEq/L.153 Regardless of the underlying mechanism, this magnitude of increase in plasma potassium concentration would be unlikely to result in detectable hyperkalemia in a recipient dog transfused with blood stored in this manner.
Table 5-1 Sodium and Potassium Concentrations of Mammalian Erythrocytes
| Species | Sodium (mEq/L) | Potassium (mEq/L) |
|---|---|---|
| Human | 10-21 | 104-155 |
| Dog LK* | 93-150 | 4-11 |
| Dog HK*121 | 54 | 124 |
| Cat | 104-142 | 6-8 |
| Horse | 4-16 | 80-140 |
| Cow LK* | 72-102 | 7-37 |
| Cow HK* | 15 | 70 |
| Sheep LK* | 74-121 | 8-39 |
| Sheep HK* | 10-43 | 60-88 |
| Swine | 11-19 | 100-124 |
HK, High potassium; LK, low potassium.
* Sheep, cattle, and dogs demonstrate polymorphism with respect to their intracellular cation concentrations, depending on the level of Na+, K+ -ATPase activity in the mature red cell membranes.
The potassium concentrations of red cells from neonatal dogs are higher than those of red cells from adult dogs.42,132,145 Red cell concentrations of potassium decrease during the first weeks of life and reach normal adult concentrations by approximately 8 to 13 weeks of age. In one study, mean red cell potassium concentrations in puppies were 19.0 mEq/L at 1 day of age, 15.1 mEq/L at 5 weeks of age, and 8.7 mEq/L at 13 weeks of age.42 Reticulocytes from adult dogs also contain higher potassium concentrations than do mature red cells.120 In adult Akitas, red cell potassium concentrations may exceed 70 mEq/L, and hemolysis results in a progressive increase in plasma potassium concentration (up to 24 mEq/L) during storage of blood.48,158
Dogs may be divided genetically into two groups based on the presence or absence of Na+, K+-ATPase activity in the membranes of their mature red cells.99,121 Dogs with red cell membrane Na+, K+-ATPase activity maintain high intracellular potassium concentrations, whereas those without red cell Na+, K+-ATPase activity maintain red cell potassium concentrations similar to those of plasma. Reticulocytes from low-potassium (LK) dogs possess Na+, K+-ATPase, but it is rapidly and completely degraded by a proteolytic process during cell maturation.100,120 Reticulocytes from high-potassium (HK) dogs have twice as much Na+, K+-ATPase activity as reticulocytes from LK dogs, but in the HK dogs, degradation of the enzyme ceases early in maturation, and sufficient activity remains in the mature red cell to account for the observed high intracellular concentration of potassium.120 The HK phenotype is inherited as an autosomal recessive trait and occurs with an incidence of 26% to 38% in the Shiba and Akita breeds in Japan and 42% in the Jindo breed in Korea.76 The HK phenotype also may be seen in the Chinese shar-pei breed and can cause pseudohyperkalemia.16 Some dogs with the HK phenotype also accumulate large amounts of reduced glutathione in their erythrocytes (so-called HK/HG phenotype), which predisposes them to oxidative injury and hemolytic anemia associated with onion ingestion.209
Red cells of English springer spaniel dogs with phosphofructokinase deficiency had potassium concentrations of 19.2 to 28 mEq/L as compared with 5.1 to 7.7 mEq/L in control dogs, and hemolytic crises in affected dogs were associated with hyperkalemia.83 The higher potassium concentration in the red cells of affected dogs was attributed in part to the large number of circulating reticulocytes (7% to 26%). The same mutation has been reported to cause phosphofructokinase deficiency and hyperkalemia in whippets.80
Hemolysis in Akitas (and presumably in other HK dogs) and thrombocytosis cause what has been called pseudohyperkalemia because these effects occur in vitro. Pseudohyperkalemia also has been reported in a dog with acute lymphoblastic leukemia before chemotherapy.95 Leakage of potassium from the leukemic cells in vitro was thought to be responsible for pseudohyperkalemia in this case. Use of plasma from small blood samples collected in an excessive volume of tripotassium ethylenediaminetetraacetic acid also may result in measured hyperkalemia.
Hypokalemia
Clinical and laboratory features
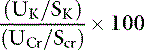
The FEK should be less than 4% for nonrenal sources of loss, and in the presence of hypokalemia, values above 4% may indicate inappropriate renal loss.60 In one study, however, FEK values for normal cats were 10.6 ± 2.1%.3 In another study of normal cats receiving a potassium-deficient diet, FEK values decreased from 10% to 12% to 3% to 6%.61 Thus, FEK values up to 6% should probably be considered normal in potassium-depleted animals with normal renal function. However, the clinical utility of FEK calculations is limited by the fact that FEK does not correlate well with 24-hour urinary excretion of potassium.3,74 The occurrence of hypokalemia in patients with metabolic alkalosis suggests vomiting of stomach contents or diuretic administration as likely causes of potassium loss. In patients with hypokalemia and metabolic acidosis, diarrhea caused by small intestinal disease, chronic renal failure, and distal renal tubular acidosis are more likely causes of potassium loss (Fig. 5-11).
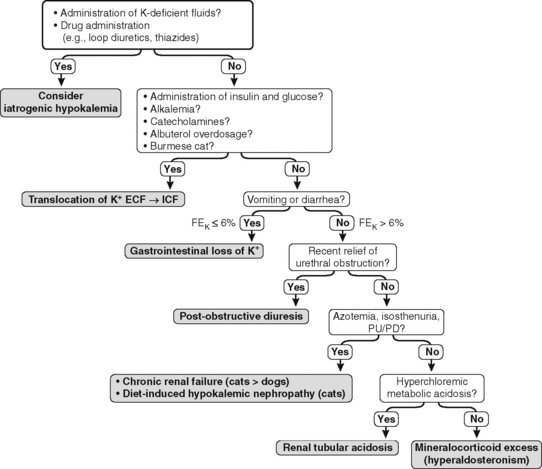
The effect of aldosterone on serum potassium excretion can also be evaluated by comparing urine and serum potassium concentrations after correcting the urine potassium concentration for reabsorption of solute-free water by the kidneys. This index has been called the transtubular potassium gradient (TTKG).39,122,204,205 A value of 5.0 or higher has been said to indicate the presence of an aldosterone effect, whereas a value of 3.0 or less is expected in the absence of mineralocorticoid activity.205 Use of the TTKG is valid only when the urine osmolality is greater than 300 mOsm/kg and the urine sodium concentration is greater than 25 mEq/L. The renal TTKG may be estimated according to the equation:
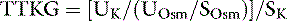
where UK is the urine potassium concentration (mEq/L), SK is the serum potassium concentration (mEq/L), UOsm is the urine osmolality (mOsm/kg), and SOsm is the serum osmolality (mOsm/kg).204,205 Values for TTKG were estimated as 3.7 ± 0.9 in normal cats and 4.2 ± 1.3 in normal dogs.52,56 Determination of TTKG was used in a dog with hypoadrenocorticism to assess the contribution of concurrent trimethoprim administration on the observed hyperkalemia.166 The causes of hypokalemia are listed in Box 5-1, and the diagnostic approach to hypokalemia is presented in Figure 5-11.



