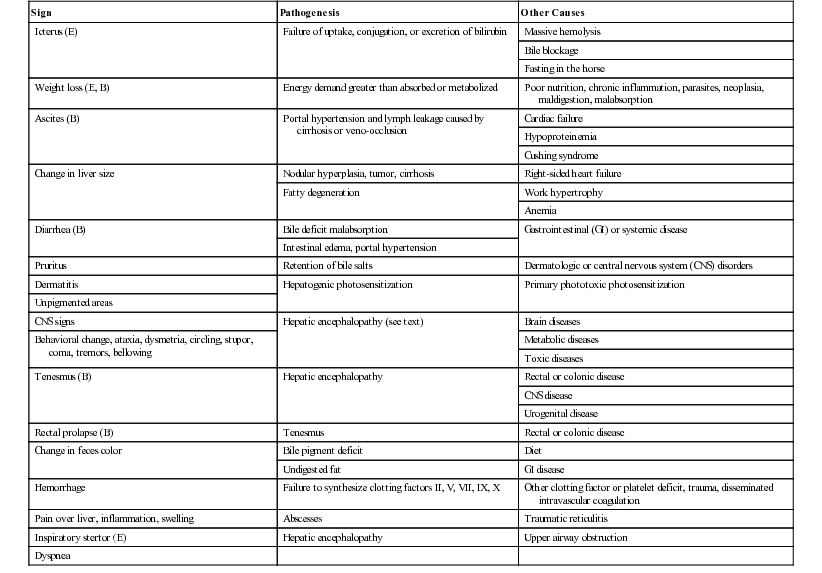
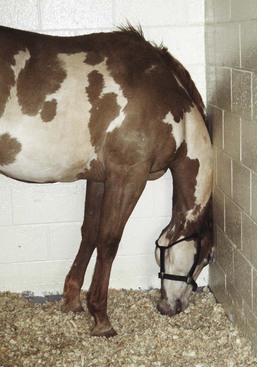
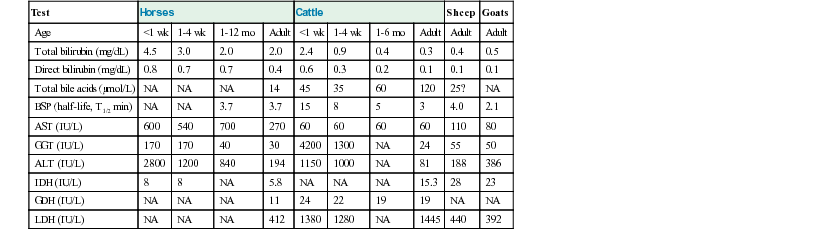
Geof W. Smith, Jennifer L. Davis, Consulting Editors Geof W. Smith In making a diagnosis, the veterinarian should first attempt to determine that the animal has liver disease and then move toward trying to identify a specific cause. Therefore, this chapter first discusses the diagnosis of liver disease, then the individual conditions causing disease in horses and ruminants. The signs of liver disease are due to failure of some of the liver’s many functions, but these signs are often not apparent early in the course of disease. Special tests may be necessary to detect early damage or minor impairment of function that has not yet produced clinical signs. As with many other organs like the heart and kidney, the liver can be diseased or damaged long before it fails to function. Early liver diseases are often inapparent to the owner or veterinarian through physical findings alone. Such cases are usually detected by finding elevated levels of liver enzymes in the serum. Pathologic changes in the liver may include biliary hyperplasia, death of hepatocytes, and fibrosis—all potentially occurring long before any signs of failure develop. Some functions may fail before others, and onset of liver failure varies with the species and disease process involved. The liver has a large reserve capacity, and close to 80% of it can be removed before regeneration and recovery are no longer possible. Regeneration can occur in areas receiving portal blood, but most cell division takes place in Rappaport zone 1 (the portal area), with cells being pushed to the central lobular area. The liver undergoes constant repair, and in humans it is estimated that hepatocytes are renewed every 50 to 75 days.1 Normal regeneration does not occur in some cases. Antimitotic agents such as pyrrolizidine alkaloid metabolites or antineoplastic drugs can prevent cell division. Regeneration may be restricted by connective tissue. Once fibrosis has bridged the various lobules, additional regeneration is impaired because the fibrosis itself perpetuates the condition. Loss of a stroma to build on or lack of portal blood supply also reduces regeneration. Many signs can be present with liver disease, but no sign is pathognomonic or present in every case. Table 33-1 lists some of the signs that may be present and other possible causes of the same signs. The clinical history is useful in some cases but not consistently. For example, liver flukes in cattle are generally restricted to certain geographic areas and occur more commonly when animals are grazing land infested with snails. Another example would be acute serum hepatitis in horses, which is known to occur most commonly in Equidae 4 to 8 weeks after administration of horse serum. In other cases the lack of specific history information may not rule in or rule out specific causes of liver disease. For example, there may not be a history of animals consuming plants containing pyrrolizidine alkaloids because of the delay between consumption and the onset of clinical signs. With the exception of change in liver size and pain over the liver elicited with pressure, most signs are related to failure of some function. Liver flukes may cause anemia and hypoproteinemia because of the effect of the parasite and its metabolites. Liver abscesses and other infections may produce signs like fever and anorexia owing to release of pyrogens and other inflammatory mediators (caused by the organism and not necessarily related to the liver itself). Icterus is typically seen in acute liver disease in horses but is not seen in many cases of chronic liver disease, and it is seen less often in ruminants unless biliary blockage occurs. Icterus is due to failure of uptake, conjugation, or excretion of bilirubin. Excess production caused by hemolysis must also be considered when icterus is present. This is likely the most common cause of icterus in ruminants and occurs with several hemolytic diseases such as anaplasmosis and copper toxicity. Horses are frequently icteric (up to 6 mg/dL unconjugated bilirubin) from anorexia or fasting even when the liver is normal. Weight loss is a common but nonspecific finding in some cases of chronic liver disease. It may be caused by anorexia or failure of metabolic functions of the liver and is probably not related to impaired fat absorption. Diarrhea is also seen, especially in cattle with chronic liver disease. It is thought to be related to portal hypertension and increased hydrostatic pressure, although the exact mechanisms are not completely understood. Diarrhea probably is not caused by fat malabsorption or steatorrhea, because the normal herbivore diet contains less than 3% fat. Ascites can also occur in animals with liver failure. The ascites results from portal hypertension caused by venous blockage producing increased hydrostatic pressure and by protein leakage into the peritoneal cavity. Production of hepatic lymph high in protein (>3 g/dL) is increased. Because the liver sinusoids are permeable to plasma proteins, the protein-containing lymph leaks into the interstitial space and then into the peritoneal cavity.2,3 Fluid moves into the abdominal cavity secondary to both osmotic and hydrostatic forces, according to Starling law. The abdominal fluid present with liver disease is a modified transudate, but the protein content may be relatively high (3 to 3.5 g/dL) owing to leakage of protein from the liver. Hypoalbuminemia can aggravate the ascites, but if it occurs alone, it usually will result in submandibular and brisket edema. Hepatic photosensitization may lead to dermatitis of the white areas, wherein the skin of these areas first becomes erythematous, then thickened with keratin crusts, and finally necrotic. This is caused by the photodynamic agent phylloerythrin, which is formed in the gastrointestinal (GI) tract of large animals by bacterial degradation of chlorophyll. After absorption into the portal circulation, phylloerythrin should be conjugated by the liver and excreted into the bile. With cholestasis, phylloerythrin may be carried to the skin, where it acts as a photodynamic agent.4 After bile duct ligation, the level of phylloerythrin steadily increases.5 Although a small amount is removed by the kidneys, the rate is not fast enough to prevent accumulation in the plasma and skin. Phylloerythrin in the skin reacts to sunlight and emits energy that causes lesions of the white areas.4 Pruritus is seen in a few cases of liver disease in horses. In humans, pruritus is assumed to be due to bile acid accumulation in the skin when it is not excreted by the liver. This same process may occur in horses, but pruritus is not usually seen in large animals with liver disease. A change in fecal color is not usually noted in either adult cattle or horses with liver disease, because other pigments like chlorophyll contribute to the color. In young animals with simple digestive tracts, much of the fecal color is from stercobilin, a metabolite of bilirubin, so with cholestasis, feces may be somewhat lighter in color. A few signs may be seen terminally in liver disease. Hemorrhage may occur when clotting factors are not synthesized in adequate amounts. Factors I, II, V, VII, IX, and X are all produced by the liver,1,3 but the disease is usually advanced before a deficit develops. Tenesmus, often followed by rectal prolapse, is seen in some cattle with liver disease. This may be associated with diarrhea, be part of hepatic encephalopathy, or be aggravated by edema of the bowel secondary to portal hypertension. Pharyngeal or laryngeal collapse with loud stertorous inspiratory noises and dyspnea has developed in some cases of hepatic failure, especially in ponies. The exact mechanism for this is not known, but it may also be part of hepatic encephalopathy.6 Horses sometimes develop a terminal hemolytic crisis caused by increased red blood cell (RBC) fragility. This has not been observed in ruminants. Hepatic encephalopathy is a neuropsychiatric syndrome caused by hepatic dysfunction or portosystemic shunting of the intestinal blood.7 It is considered a potentially reversible metabolic or neurotransmitter disorder, but it is associated with characteristic (although nonspecific) lesions in the central nervous system (CNS), such as altered astrocytes. Signs of hepatic encephalopathy are often subtle and nonspecific. Behavioral changes may be detected by the owner, who is more familiar with the patient’s normal activity. Some docile animals become excitable and difficult to control, whereas other normally unruly animals may become passive. Depression and incoordination are frequent manifestations, and some animals may walk aimlessly or even head press (Fig. 33-1).8 Apparent blindness is seen in some horses, and foot stomping was reported in 7 of 25 cases.9 These animals eventually develop a stupor and may end up in hepatic coma. Yawning may be seen in horses, and ponies may have a stertorous respiratory noise. Ruminants show signs of tenesmus and sometimes vocalize excessively. In humans, hepatic encephalopathy is diagnosed by neuropsychological tests, which are not possible in animals.10 The pathophysiology of hepatic encephalopathy remains undefined and is controversial.8 It occurs when portal blood bypasses the liver, as with congenital shunts in dogs, or with shunts secondary to portal hypertension induced by alcoholic cirrhosis in humans, or when the blood goes through an inadequately functioning liver. How neurologic function is altered has not been determined, but speculation abounds. It seems possible to incriminate synergistic neurotoxins that bypass the liver. Most will agree that ammonia levels play a central role in pathogenesis. Blood ammonia is elevated in most cases of hepatic encephalopathy because the liver is not metabolizing ammonia to urea. Encephalopathy can be precipitated in cirrhotic patients by adding ammonia-generating substances, and there is an increase in cerebrospinal glutamine, the product to which ammonia is detoxified. Astrocytes contain the enzyme glutamine synthetase, which adds a molecule of glutamate to ammonia to form glutamine. The amount of glutamine formed in the brain correlates with the severity of hepatic encephalopathy.11 However, much higher concentrations of ammonia are needed to induce coma in normal animals, and there is poor correlation between ammonia levels and degree of encephalopathy.9 Cases of hyperammonemia and encephalopathy without liver disease have been reported in horses.12 Magnetic resonance imaging (MRI) of human cirrhotic patients reveals increased image intensities from the globus pallidus region, probably caused by manganese deposits.10 Serum manganese is increased in these patients, which correlates with the increased MRI intensities and negatively with neuropsychological tests. This would indicate that manganese is another potential toxin that can enter the brain if not effectively removed from the blood by the liver. There may be an imbalance of true inhibitory and excitatory neurotransmission. γ-Aminobutyric acid (GABA) is an inhibitory neurotransmitter. The increase in GABAergic tone was originally thought to result from an increase in GABA or its receptor sites. More recently, it has been proposed that neurosteroids with GABA-agonist properties could be involved.13 Ammonia is believed to play a role in the metabolism of GABA in the brain and thus could act synergistically.14 Also, receptor sites for benzodiazepine are increased in the brain of animals with hepatic encephalopathy.15 Benzodiazepine augments the activity of GABA in stimulating the inhibitory neuron and causing sedation. False neurotransmitters have been proposed to cause the abnormal nerve function. Increased amounts of tryptophan, phenylalanine, and tyrosine in the brain could cause more serotonin to accumulate in the brain. Plasma amino acid ratios are altered in horses with pyrrolizidine alkaloid–induced liver disease.15 Concentrations of branched-chain amino acids (valine, isoleucine, leucine) are decreased, and concentrations of aromatic amino acids (tyrosine, phenylalanine, free tryptophan) are increased, partially because the liver is not adequately metabolizing aromatic amino acids. Amino acids are transported across the blood-brain barrier by a common transport system, so they compete for entry into the CNS. It has been suggested that higher levels of aromatic amino acids in the CNS would lead to formation of increased amounts of inhibitory neurotransmitters or to alteration of catecholamine or monoamine neurotransmitters like GABA or l-glutamate. A number of enzymes are compartmentalized in the hepatocyte or in bile duct epithelium. This compartmentalization is useful in holding insoluble molecules close to the enzymes for chemical reactions. Hepatocyte damage may result in release of the enzymes into the circulation, and cholestasis may cause increased release from bile epithelium. Serum levels of these enzymes may therefore be an indication of hepatocyte integrity or bile excretion. Table 33-2 lists frequently tested enzymes and their characteristics. Serum concentrations for some of these enzymes vary with the age of the animal and sometimes even with type or use. Table 33-3 lists some approximate upper limits of normal for animals of various ages. Normal values, especially for the enzymes, should be established by each laboratory. TABLE 33-2 Liver-Derived Enzymes * Elevated in urine, not blood. ALP, Alkaline phosphatase; ALT, alanine aminotransferase (formerly SGPT, serum glutamic-pyruvic transaminase); AST, aspartate aminotransferase (formerly SGOT, serum glutamic-oxaloacetic transaminase); GGT, γ-glutamyltransferase; GDH, glutamate dehydrogenase; IDH, l-iditol dehydrogenase (formerly SDH, sorbitol dehydrogenase); LDH, lactate dehydrogenase; OCT, ornithine carbamoyltransferase. The cytosol of the hepatocyte contains high activities of the enzymes aspartate aminotransferase (AST, formerly serum glutamic-oxaloacetic transaminase [SGOT]), l-iditol dehydrogenase (IDH, formerly sorbitol dehydrogenase [SDH]), ornithine carbamoyltransferase (OCT), glutamate dehydrogenase (GDH), and lactate dehydrogenase (LDH). These are generally referred to as leakage enzymes, since with acute or chronic hepatocellular injury or necrosis the activity of these enzymes increases because they essentially “leak” out of hepatocytes into the serum.16 Cholestasis increases production of other enzymes such as γ-glutamyltransferase (GGT) and alkaline phosphatase (ALP). Activity of these enzymes generally increases secondary to biliary obstruction caused by conditions like liver fluke infection or cholelithiasis. The cholestatic enzymes are more likely to have high serum concentrations in chronic hepatic disease than the leakage enzymes because of bile duct constriction or obstruction secondary to fibrosis.16 GGT also is present in pancreas, mammary gland, lung, kidney tubules, and other duct epithelium, but serum levels usually are not elevated with renal disease, because the enzyme is lost in the urine. Serum concentrations are normally higher in neonatal calves (sometimes >4000 IU/L after suckling)17 because the enzyme is concentrated in colostrum.18,19 GGT levels are also higher in foals than in adult horses, but the higher concentrations may partly be the result of increased production.20 GGT is elevated in many horses with right dorsal displacement of the large colon, possibly caused by transient extrahepatic bile duct obstruction.21 Elevated GGT is the most sensitive indicator of liver disease in the horse, and levels will remain elevated for several weeks.9,22 ALP is often elevated in chronic liver disease of the horse but is variable in ruminants. ALP can come from other sources like bone, intestines, placenta, and macrophages, so it is not specific. Both ALP and GGT are elevated with cholestasis.23 The dehydrogenases (i.e., IDH [SDH], LDH, GDH) are found in hepatocytes and are elevated with acute hepatocyte damage, but serum concentrations may return to normal or below normal in chronic liver disease. IDH is liver specific and extremely useful in detecting active hepatocellular necrosis, but it is not as stable as some of the enzymes (levels drop significantly after 24 hours of refrigeration). Increased IDH activity can be seen in ruminants with infectious, inflammatory, toxic, or metabolic insults to the liver. In cases of chronic disease, however, the IDH concentration may be normal because either too few hepatocytes are damaged or the hepatocellular mass is substantially reduced.24 AST and LDH are found in many tissues other than liver (especially muscle), so they are nonspecific unless isoenzymes are determined. Muscle injury or necrosis, especially in recumbent animals, will often result in marked increases in both AST and LDH. The clinician must evaluate AST and LDH concentrations in conjunction with muscle-specific enzymes like creatine kinase (CK). Elevations in these enzymes together with increased CK activity generally suggests muscle injury. However, AST elevations occurring with CK values that fall within the reference range gives a strong suggestion of hepatic disease. AST and LDH concentrations are also found in erythrocytes, so if the serum is severely hemolyzed or allowed to remain on the clot for an extended period, falsely elevated values from leakage from red blood cells will result. Ammonia is produced in the GI tract from the digestion of proteins and amino acids, absorbed by the intestine into blood, and carried to the liver in the portal circulation. Ammonia is converted to urea in the liver by the hepatic urea cycle. Ammonia is a useful test for assessing liver function, but it is generally not included in most serum biochemistry panels because it is highly volatile and requires special sample handling. Serum urea nitrogen (SUN) may also have some value as an indicator of hepatic disease in ruminants, since urea is produced in the liver. In severe cases of liver failure, SUN is usually low and ammonia often high. However, since rumen bacteria synthesize protein from urea, ruminants that have been off feed have low SUN concentrations because all the urea is being used by rumen bacteria for protein production. Thus low SUN levels are not always specific for hepatic disease. No single liver enzyme value can consistently confirm or rule out the presence of liver disease. A combination of tests, including serum enzymes, total bile acids, ultrasound, and liver biopsy, may be needed. Some blood constituents may be altered because of failure of certain metabolic functions of the liver, but none of these changes is specific for liver disease. Blood glucose is sometimes slightly lower in severe liver disease, especially in young animals, possibly because of decreased gluconeogenesis, but none of 28 adult cases in one study had low blood glucose concentrations, and in fact, 14 cases had hyperglycemia.9 In some toxic liver diseases, blood ammonia levels can increase fourfold or more because the urease necessary to convert ammonia to urea is found only in the liver. For the same reason, SUN may decrease, especially in the terminal phases. In the later stages, blood-clotting factors may be diminished, with delayed partial thromboplastin time (PTT) and other clotting times. Terminally, serum albumin concentration may decrease. Considerable protein synthesis takes place in the liver, which produces all the plasma proteins except γ-globulins. The amount of these proteins in the blood, however, depends not only on the rate of synthesis but also on the rate of removal. The albumin half-life in cattle is about 16.5 days; in the horse, 19.4 days; and in sheep, 14 days.25 Therefore, serum albumin is reduced mainly in chronic liver disease. With liver damage, the synthesis of α-globulins and β-globulins is increased, so total plasma protein concentration is invariably normal or elevated, but the albumin/globulin (A/G) ratio may be decreased. The healthy liver has a large reserve for protein synthesis, and lost protein can be replenished. In one study, 18% of the horses with chronic liver disease and 6% of those with acute liver disease had albumin concentrations below the reference value.26 In a British study, only 6 of 37 liver disease cases were hypoalbuminemic, and none of these was hypoproteinemic.9 Therefore, it is assumed that hypoalbuminemia is not a common feature in horses with liver disease, and that hypoproteinemia is rare. Amino acid ratios are altered in liver disease; short branched-chain amino acids are decreased, whereas aromatic amino acids are increased.27 Because the liver excretes a number of endogenous compounds and foreign substances injected into the animal, the rate of excretion or clearance of these substances can be used to test the excretory function of the liver. Bilirubin itself can be used and, if elevated above normal, would indicate liver failure, bile blockage, or excess production from hemolysis. In the horse, bilirubin is also increased during fasting in animals without liver disease.28 With liver damage in the horse or ruminant, most of the retained bilirubin is unconjugated (indirect reacting), and the direct-to-total ratio is usually less than 0.3. With bile blockage or intrahepatic cholestasis, the direct-to-total ratio may be greater than 0.3 in the horse or 0.5 in cattle. Bilirubin is the main bile pigment and is produced from heme, 75% of which comes from RBCs. When the erythrocytes are broken down, heme is converted first to biliverdin and then to bilirubin in the macrophage system. This unconjugated bilirubin is insoluble and must be bound to albumin for transfer; the kidneys do not remove this bound bilirubin. Unconjugated bilirubin is taken up by the hepatocytes with cytosolic binding proteins within the hepatocyte. In the hepatocyte, some bilirubin is conjugated to diglucuronide, but in the horse, more than half the bilirubin in bile is conjugated with glucose. Conjugated (direct-reacting) bilirubin is water soluble, and some will enter the general circulation; if concentration is sufficiently high, the kidneys will filter it into the urine. Conjugated bilirubin is secreted into the bile canaliculi by an energy-dependent transport process. In most species, this is the rate-limiting step, but this may not be true in large herbivores. Conjugated bilirubin passes into the intestine through the bile ducts, and if they are blocked, both conjugated and unconjugated bilirubin will increase in the plasma. In the intestinal tract, bilirubin is converted to urobilinogen by anaerobic bacteria. Some urobilinogen is absorbed and reexcreted by the liver, but a small fraction will pass the normal liver and be excreted in the urine. With complete biliary blockage, there will be no urobilinogen in the urine, and with hemolysis, there may be increased urobilinogen in the urine. Urobilinogen is not very stable in the urine, so analysis must be done within 1 or 2 hours or the amount detected will be erroneously low. Fasting decreases the efficiency of plasma bilirubin removal in all species, but the horse shows a greater rise in plasma bilirubin, often reaching a plateau two to three times the normal state. This increase is due to a decrease in removal of bilirubin by the hepatic transport and not by an increase in its production.28,29 In normal ruminants, total bilirubin concentrations are low compared to other species, and the magnitude of increase is generally small and inconsistent even with severe liver damage.2 Ruminants with cholestatic disease may have moderate increases in serum bilirubin, but this is not a consistent finding. Increases in direct bilirubin are usually the result of hemolysis. Serum total bile acid concentration is a good test of liver function in horses. The concentration of bile acids in the serum will be increased if there is hepatocyte damage, blockage of bile flow, or shunting of portal blood to the systemic circulation, bypassing the liver. The liver synthesizes bile acids from cholesterol. Cholic and chenodeoxycholic acids are the primary bile acids that are conjugated with amino acids before excretion into the bile. Only conjugated bile acids are present in the intestine; they are soluble and form micelles with fat because of their detergent properties. Most bile acids excreted in bile are resorbed by an active transport system in the ileum and carried by the portal circulation back to the liver for reexcretion. In most species, more than 95% of the bile acids are resorbed and recirculated through the enterohepatic circulation.30 Many simple-stomached animals have a postprandial increase in serum bile acid because bile is released from the gallbladder during eating and subsequently resorbed by the ileum. This does not seem to be important in the horse, which has no gallbladder, or in cattle, in which no relationship to feeding could be found.31 Table 33-3 lists the upper limits of normal for liver function tests in large animals of various ages. In cattle there is an hour-to-hour fluctuation that could be as much as 60 µM/L, along with considerable variation between individual cattle.31,32 In dairy cattle a significant decrease in serum bile acid concentrations occurs throughout the course of lactation.33 In the horse, serum concentration above 14 µM/L would indicate liver damage, bile blockage, or shunting.33 Serum total bile acid concentration is likely a more sensitive indicator of liver disease in the horse than serum bilirubin; only 8 of 34 horses with liver disease had elevated bilirubin, but 31 of 37 had elevated bile acids.9 In adult cattle, because of the hour-to-hour variations, bile acid concentration on a single sample would have to be above 126 µM/L in beef cattle and 88 µM/L in dairy cattle to be an indication of liver disease.34,35 Although ruminants have a large normal range for bile acid concentrations, significant increases have been shown to be a specific indicator of hepatic disease.35 A number of dyes are excreted primarily by the liver; sulfobromophthalein (bromsulphalein [BSP]) is used most often. In large animals, BSP clearance (half-time) is used much more than the retention test. In the test, 500 to 1000 mg (≈2 mg/kg) of BSP is injected intravenously. Blood samples are taken before the injection and two to four times 5 to 12 minutes after the injection (i.e., at 5, 7, 9, and 11 minutes). These samples are analyzed for color produced by BSP, and a half-life is determined by plotting the points on semilog paper. Normal horses have a BSP half-life of less than 3.5 minutes, and for ruminants it is less than 5 minutes. This may be increased in very young animals (see Table 33-3) and in pregnant females. Other dyes, such as indocyanine green, have some advantage (less renal excretion than BSP) but at present are too costly for use in large animals.
Diseases of the Hepatobiliary System

Diagnosis of Liver Disease*
Liver Diseases versus Liver Failure
Liver Reserve and Regeneration
Signs of Liver Disease and Pathophysiology
Hepatic Encephalopathy
Laboratory Tests and Liver-Derived Serum Enzymes
Enzyme
Specificity
Problems
GGT
Liver
High in young animals from colostrum
Kidney*
Pancreas
ALP
Liver
Not specific
Bone
Intestine
Macrophages
Placenta
IDH (SDH)
Liver
Not elevated in chronic disease; short life; not stable
GDH
Liver
Not elevated in chronic disease
AST (SGOT)
Liver
Not specific
Muscle
Heart
ALT (SGPT)
Liver
Low concentration in cattle and horses; not a good indicator in large herbivores
LDH
None unless isoenzymes
Not elevated in chronic disease; short life; not specific
OCT
Liver
Analysis not routinely available
Excretion Tests for Liver Function
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Diseases of the Hepatobiliary System
Chapter 33