Chapter 139 This chapter provides a rational approach to the diagnosis of hepatobiliary disease in small animal patients. The diagnosis of a primary hepatobiliary disorder can be a challenge; therefore a logical approach is necessary, incorporating all aspects of the case from signalment to diagnostic imaging and histopathology. The clinical signs of hepatobiliary disease generally reflect deficiencies in the varied functions of the liver. These diverse metabolic and biochemical activities include carbohydrate, lipid, and protein metabolism; fat digestion; detoxification of endogenous and exogenous substances; and immune surveillance. However, the clinical signs that develop with liver disease are seldom specific for that organ. Furthermore, the high blood flow of the liver, its dual blood supply (systemic and portal), and its role in detoxification render the liver sensitive to injury from systemic disorders and diseases in organs drained by the portal circulation (Box 139-1). To complicate the situation, the tremendous hepatic reserve capacity makes the appearance of relatively specific signs of hepatobiliary dysfunction such as icterus, hypoglycemia, bleeding tendencies, hepatic encephalopathy (HE), and ascites occur only in late-stage disease. The first step in the diagnosis of hepatobiliary disease is to obtain an accurate history. Pertinent information includes the use of potentially hepatotoxic drugs, supplements, or nutraceuticals; exposure to environmental toxins, infectious agents, or recent anesthetic events; and details on housing, supervision outdoors, and travel and vaccine status (leptospirosis, canine adenovirus). Recognition of an agent’s hepatotoxic potential (Box 139-2) and prompt withdrawal can prevent further liver damage (see Chapters 140 and 141). Often a sequence of events may increase the suspicion for hepatobiliary disease. A history of inappetence and weight loss in a previously over-conditioned feline is suggestive of hepatic lipidosis. Anesthetic intolerance, failure to thrive, and postprandial behavioral abnormalities in a predisposed canine breed should increase suspicion of a portosystemic vascular anomaly (PSVA) (Berent and Tobias, 2009). Primary hepatobiliary disease always should be considered in breeds predisposed to inflammatory/fibrotic hepatopathies (Box 139-3). Alanine aminotransferase (ALT), aspartate aminotransferase (AST), alkaline phosphatase (ALP), and γ-glutamyl transferase (GGT) are serum enzymes used as screening tests for hepatobiliary disease (Center, 2007). Increases occur as a result of (1) leakage from damaged hepatobiliary cells (ALT, AST); (2) elution from damaged membranes (ALP, GGT); or (3) increased synthesis (ALP). Although elevations in these serum enzymes have a high sensitivity for the detection of hepatobiliary damage, increases also occur in the absence of clinically important primary hepatobiliary disease. This discordance occurs for several reasons. First, increases in serum hepatobiliary enzymes can originate from nonhepatic tissues. Second, in the dog endogenous or exogenous corticosteroids and phenobarbital can induce the production of excess hepatobiliary enzymes in the absence of liver damage. Finally, the liver is uniquely susceptible to secondary injury from primary disease in other organs, particularly the pancreas and gastrointestinal tract (see Box 139-1). ALP is a membrane-bound enzyme (see Web Chapter 50). In dogs ALP has a high sensitivity (80%) but low specificity (51%) for hepatobiliary disease. Its low specificity is caused by the presence of several isoenzymes and its sensitivity to drug induction. In the dog three isoenzymes make up the total serum ALP (T-ALP), including a bone (B-ALP), liver (L-ALP), and corticosteroid (C-ALP) isoenzyme. B-ALP makes up one third of the T-ALP in dogs and is elevated in conditions with increased osteoblastic activity such as bone growth, osteomyelitis, osteosarcoma, and secondary renal hyperparathyroidism. L-ALP is present on the luminal surface of biliary epithelial cells and the hepatocyte canalicular membrane. L-ALP Idiosyncratic hepatotoxic reactions to phenobarbital may occur in dogs, leading to chronic inflammatory disease or the hepatocutaneous syndrome. In both of these disorders moderate-to-marked increases in ALP, moderate increases in ALT, and mild increases in AST typically are seen. Phenobarbital therapy in the dog has been reported to induce the production of hepatobiliary enzymes (primarily ALP). Prospective studies to assess the relative role of induction versus damage in dogs on phenobarbital therapy have resulted in conflicting results. One study demonstrated no increase in ALT and ALP enzyme activity in whole-liver homogenates from phenobarbital-treated dogs, suggesting that induction was not occurring (Gaskill et al, 2005). However, another study found increased T-ALP activity in the liver of phenobarbital-treated dogs, supportive of induction (Unakami et al, 1987). Overall the available literature suggests that mild-to-moderate increases in ALP (up to five times the upper limit of normal) and ALT (usually less than two times the upper limit of normal) may reflect enzyme induction. However, increases in GGT and AST seldom are caused by induction and may be suggestive of primary liver disease. Hepatobiliary disease may result in ascites formation secondary to portal hypertension (Buob et al, 2011). Portal hypertension occurs as a result of obstruction at the level of the right atrium/cranial vena cava, hepatic parenchyma, or portal vein. Portal hypertension from the latter two causes typically results in a low-protein ascites, whereas posthepatic portal hypertension is associated with a high-protein ascites. Generally ascitic fluid caused by hepatic disease is a pure transudate but with chronicity may have characteristics of a modified transudate. An effusion bilirubin value in excess of serum bilirubin is consistent with bile duct/gallbladder rupture. Ascites is rare even in cats with end-stage liver disease. In cases in which secondary hepatobiliary disease is suspected, assessment for pancreatitis (serum lipase levels/pancreatic lipase immunoreactivity) and evaluation for an underlying endocrinopathy (hypothyroidism, hyperthyroidism, hyperadrenocorticism, and adrenal hyperplasia syndromes) or gastrointestinal disease (inflammatory bowel disease) may be pursued (see Box 139-1).
Diagnostic Approach to Hepatobiliary Disease
Laboratory Evaluation of Hepatobiliary Disease
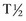 is 70 hours in dogs and 6 hours in cats. Because of the cats’ short
is 70 hours in dogs and 6 hours in cats. Because of the cats’ short 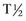 and the fact that feline hepatocytes contain less ALP, increases typically do not approach those seen in canine patients. These characteristics and the fact that cats lack C-ALP make T-ALP less sensitive (50%) but more specific (93%) for liver disease than in the dog. The largest increases in L-ALP are associated with focal or diffuse cholestatic disorders and primary hepatic neoplasms. Less dramatic increases are found in chronic hepatitis, hepatic necrosis, and canine nodular hyperplasia. C-ALP is located on the hepatocyte canalicular membrane. C-ALP increases in dogs exposed to exogenous corticosteroids or in cases of spontaneous hyperadrenocorticism; however, increases also are associated with chronic illness, possibly secondary to increases in endogenous glucocorticoid secretion.
and the fact that feline hepatocytes contain less ALP, increases typically do not approach those seen in canine patients. These characteristics and the fact that cats lack C-ALP make T-ALP less sensitive (50%) but more specific (93%) for liver disease than in the dog. The largest increases in L-ALP are associated with focal or diffuse cholestatic disorders and primary hepatic neoplasms. Less dramatic increases are found in chronic hepatitis, hepatic necrosis, and canine nodular hyperplasia. C-ALP is located on the hepatocyte canalicular membrane. C-ALP increases in dogs exposed to exogenous corticosteroids or in cases of spontaneous hyperadrenocorticism; however, increases also are associated with chronic illness, possibly secondary to increases in endogenous glucocorticoid secretion.
Ancillary Diagnostic Tests
![]()
Stay updated, free articles. Join our Telegram channel
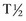 ) of ALT is
) of ALT is 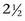 days in dogs. No published values are available for cats; however, the
days in dogs. No published values are available for cats; however, the 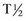 is presumed to be much shorter (around 6 hours). The largest elevations in ALT occur with acute hepatocellular necrosis and inflammation. Mild-to-moderate elevations occur with primary hepatic neoplasia. AST, which is present in the cytosol and mitochondria, is more sensitive but somewhat less specific for liver disease than ALT. Increases in AST typically parallel those of ALT but are of a smaller magnitude. AST elevations in excess of ALT indicate either a muscle source or the release of mitochondrial AST caused by severe irreversible hepatocellular injury. The
is presumed to be much shorter (around 6 hours). The largest elevations in ALT occur with acute hepatocellular necrosis and inflammation. Mild-to-moderate elevations occur with primary hepatic neoplasia. AST, which is present in the cytosol and mitochondria, is more sensitive but somewhat less specific for liver disease than ALT. Increases in AST typically parallel those of ALT but are of a smaller magnitude. AST elevations in excess of ALT indicate either a muscle source or the release of mitochondrial AST caused by severe irreversible hepatocellular injury. The 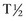 of AST is 22 hours in the dog and 77 minutes in the cat.
of AST is 22 hours in the dog and 77 minutes in the cat.
Full access? Get Clinical Tree


