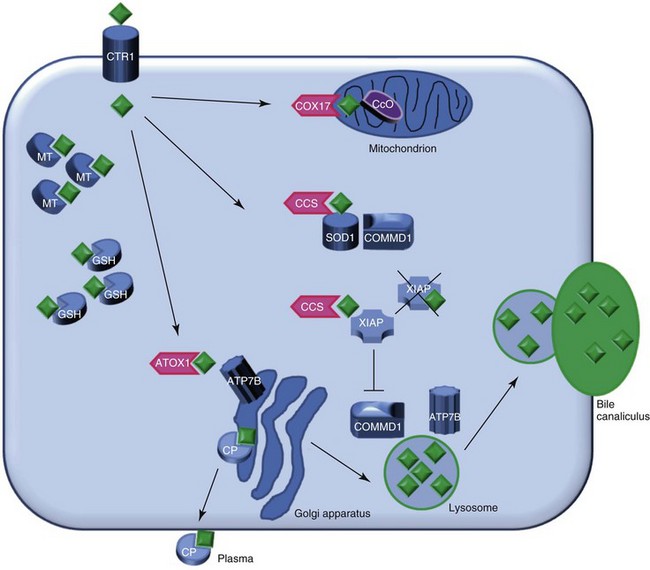
Web Chapter 48 Copper metabolism is a complex process that is regulated on cellular and organ levels (Web Figure 48-1). Copper uptake takes place in the small intestine, where its absorption is mediated via copper transporter 1 (CTR1). The P-type ATPase ATP7A shuttles copper via the basal side of the enterocytes into the portal circulation. In the blood, small molecules such as histidine and serum proteins such as macroglobulin and albumin bind copper for transportation to the liver. CTR1 mediates hepatocellular uptake of copper. In the cytosol, copper is sequestered immediately by small molecules such as metallothionein and glutathione. Specialized copper chaperones shuttle copper to their destination proteins. The copper chaperone for superoxide dismutase (CCS) shuttles copper to superoxide dismutase (SOD1), a protein involved in oxidative stress defense and to XIAP, the X-linked inhibitor of apoptosis. Binding of copper to XIAP leads to an increased intracellular degradation of XIAP, which causes a lowering of the apoptotic threshold. COX17 is the chaperone for cytochrome C oxidase, which plays an important role in cellular respiration and in the mitochondria. ATOX1 delivers copper to ATP7B, which is necessary for copper incorporation into ceruloplasmin (CP) and facilitates copper excretion in bile. Mutations of ATP7B result in the human form of copper toxicosis: Wilson’s disease. COMMD1 can interact with ATP7B and works in conjunction with this protein in the process of copper excretion into bile. Other roles for COMMD1 in copper metabolism are under investigation and include a role in maturation and activation of SOD1 and the stabilization of ATP7A. COMMD1 function itself is in part regulated by XIAP. Web Figure 48-1 Model of hepatocyte copper metabolism. Copper (diamonds) enters the cell via copper transporter 1 (CTR1) and is sequestered in the cytoplasm by the small molecules metallothionein (MT) and glutathione (GSH). Transportation of copper to the destination molecules takes place via copper chaperones. COX17 shuttles copper to the cytochrome C oxidase (CcO) in the mitochondria. CCS is the chaperone for superoxide dismutase (SOD1). Recently COMMD1 was shown to interact with SOD1, and this interaction requires CCS-mediated copper incorporation in SOD1. ATOX1 transports copper to ATP7B in the trans-Golgi network, where incorporation of copper in apo-ceruloplasmin (CP) takes place. Holo-ceruloplasmin subsequently is excreted in the plasma. The precise mechanism for export of excess copper in the bile is not resolved completely, but it is hypothesized that ATP7B and COMMD1 mediate fusion of copper-loaded vesicular compartments to the apical membrane. Furthermore, COMMD1 may play a role in the maintenance of ATP7B. XIAP can inhibit COMMD1 by promoting its degradation, resulting in cellular copper accumulation. XIAP can receive copper from CCS, and copper binding of XIAP results in its degradation and decrease in caspase inhibition, which may result in enhanced apoptosis. (Reprinted with permission from Mammalian Genome.)
Copper-Associated Hepatitis
Copper Metabolism
< div class='tao-gold-member'>
Chapter 48: Copper-Associated Hepatitis
Only gold members can continue reading. Log In or Register to continue

Full access? Get Clinical Tree


