Chapter 87 Carbon Monoxide
PATHOPHYSIOLOGY
Hypoxic Toxicity
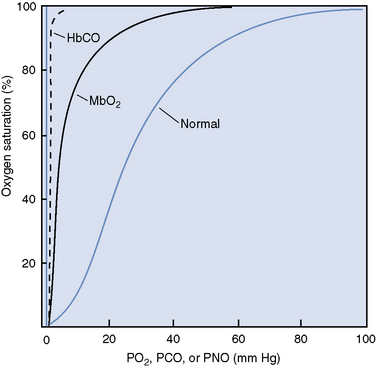
Carbon monoxide competes with oxygen for hemoglobin binding sites with 200 to 250 times the affinity.1,16 Carbon monoxide binds two of the four available heme groups in each molecule of hemoglobin, causing a decrease in the oxygen carrying capacity of 50% (Figure 87-1) and shifting the oxyhemoglobin dissociation curve down.16,17 Thus very low levels of carbon monoxide in the blood result in markedly reduced oxygen carrying capacity despite a normal hemoglobin concentration and normal partial pressure of oxygen.
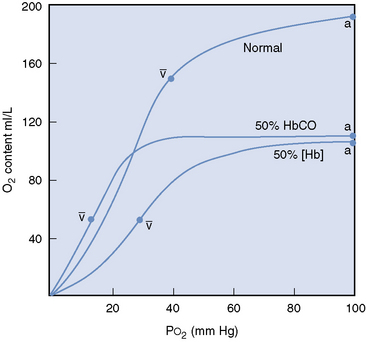
The dissociation of oxygen from hemoglobin is also affected by carbon monoxide, which binds tightly to hemoglobin, markedly disturbing the chemical equilibrium of the molecule.16,17 The change in equilibrium of the COHb results in an interference with both the association and dissociation of oxygen.16 Therefore oxygen bound to hemoglobin, which is also bound to carbon monoxide is not easily released to the tissues, and the oxyhemoglobin dissociation curve is shifted to the left (Figure 87-2).16,17 Decreased release of oxygen to the tissues exacerbates the cellular hypoxia caused by decreased oxygen content and subsequent delivery.