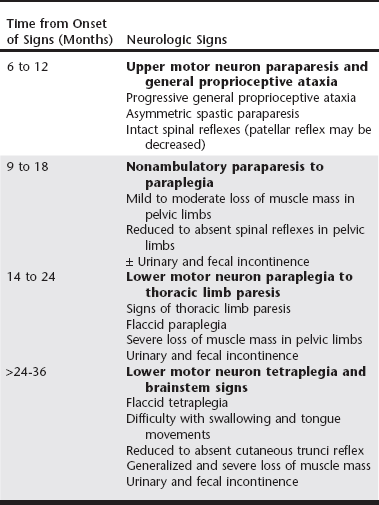
Chapter 234 Canine degenerative myelopathy (DM) was first described as an insidious, progressive general proprioceptive ataxia and upper motor neuron (UMN) spastic paresis of the pelvic limbs ultimately leading to paraplegia and necessitating euthanasia (Averill, 1973). Although most of the dogs in the initial reports were German shepherd dogs, other breeds were represented (Averill, 1973; Braund and Vandevelde, 1978; Griffiths and Duncan, 1975). DM is now recognized as a common problem in a number of breeds, with an overall prevalence of 0.19% (Coates et al, 2007; Coates and Wininger, 2010). Additionally, the clinical spectrum of DM has been broadened to encompass both the UMN and lower motor neuron (LMN) systems. Discovery of a missense mutation in the superoxide dismutase 1 gene (SOD1) provided further understanding that this canine disease may share pathogenic mechanisms with some forms of human amyotrophic lateral sclerosis (ALS, or Lou Gehrig’s disease) (Awano et al, 2009). Since it was first described by Averill (1973), canine DM, because of its histopathologic features, was termed a nonspecific degeneration of spinal cord tissue of undetermined cause. Awano and colleagues (2009) identified a c.118G>A transition of glutamate to lysine in SOD1 that predicted an E40K missense mutation underlying canine DM. In this initial study there was a highly significant association between homozygosity for the SOD1:c.118A allele and the DM phenotype in Pembroke Welsh corgis and also in four other dog breeds: boxer, Chesapeake Bay retriever, German shepherd dog, and Rhodesian ridgeback. SOD1 is a ubiquitous intracellular protein functioning as a free radical scavenger. Mutations in SOD1 are known to cause some forms of familial ALS in humans. ALS, the most common adult motor neuron disease, is characterized by loss of motor neurons causing stiffness and slowing of muscle movements, difficulty speaking and swallowing, muscle atrophy, and severe weakness. The Greek word amyotrophy means “muscles without nourishment.” Lateral is the location within the spinal cord of axonal disease, and sclerosis refers to replacement of diseased axons by sclerotic or “scar” tissue. In addition, Awano and associates (2009) demonstrated that, as in ALS, cytoplasmic aggregates that bind anti-SOD1 antibodies were present in the spinal cords of dogs with DM that were homozygotic for SOD1:c.118A. The aggregates are thought to form because amino acid substitutions force SOD1 to assume an unstable conformation. It is unclear if the aggregates cause or contribute to the neurodegeneration or are a by-product of other neurodegenerative processes. No similar SOD1 antigen–containing aggregates are found in healthy dogs lacking known SOD1 mutations. Not all SOD1:c.118A homozygotes develop clinical signs; therefore DM appears to be an incompletely penetrant autosomal-recessive disease, whereas most human SOD1 mutations are autosomal dominant. Thus homozygosity for the E40K mutation is a major risk factor for canine DM. We have detected the DM-associated SOD1:c.118A allele in over 100 different dog breeds. The mutant allele appears to be very common in some breeds. It remains to be seen whether or not homozygosity for the mutation puts dogs of all of these different genetic backgrounds at risk of developing DM. Recently another mutation in SOD1 has been discovered in a DM-affected Bernese mountain dog (Wininger et al, 2011). This finding serves as a reminder that direct DNA tests can indicate the presence or absence of disease-causing alleles but cannot be used to rule out a diagnosis because other sequence variants in the same gene or in a different gene might produce a similar disease phenotype. Subsequent to the discovery of the SOD1 mutation underlying canine DM, the clinical spectrum became more clearly defined with identification of the stages of disease progression (Table 234-1). Dogs with DM follow a predictive pattern of clinical signs that begins with UMN pelvic limb paresis and general proprioceptive ataxia, progresses to LMN paraparesis, and then spreads to involve the thoracic limbs and brainstem. The earliest clinical signs appear when dogs are at least 8 years or older, and the mean age of onset is 9 years. There is no sex predilection. The clinical course of DM can vary after the presumptive diagnosis is made; the mean disease duration is 6 to 12 months in larger dog breeds, at which point dogs become nonambulatory paraparetics. Pet owners usually elect euthanasia when their dogs can no longer support weight with their pelvic limbs. Dogs of smaller breeds can be cared for by the pet owner over a longer time; for example, the median disease duration in the Pembroke Welsh corgi was 19 months (Coates et al, 2007). DM-affected Pembroke Welsh corgis often have signs of thoracic limb paresis at the time of euthanasia. TABLE 234-1 Neurologic Signs in Dogs with Degenerative Myelopathy Based on Disease Progression Note: Shading represents the disease stages at which lower motor neuron signs are present. The earliest clinical signs of DM are general proprioceptive ataxia and mild spastic paresis in the pelvic limbs. Worn nails and the appearance of asymmetric pelvic limb lameness can be seen on physical examination. Asymmetry of signs at disease onset is reported frequently. At disease onset, spinal reflex abnormalities are consistent with UMN paresis localized in the T3 to L3 spinal cord segments. Patellar reflexes may be normal or exaggerated to clonic; however, hyporeflexia of the patellar reflex also has been described in dogs at a similar disease stage (Griffiths and Duncan, 1975). Involvement of the dorsal roots of the femoral nerve may inhibit sensory impulses from stretch receptors located in the quadriceps muscle. Flexor (withdrawal) reflexes also may be normal or show crossed extension (suggestive of chronic UMN dysfunction). Often within 6 to 12 months from the time of disease onset, dogs progress to nonambulatory paraparesis. If the DM-affected dog is not euthanized early, clinical signs will progress to LMN paraplegia and ascend to affect the thoracic limbs within 18 to 24 months. LMN signs emerge as hyporeflexia of the patellar and withdrawal reflexes, flaccid paralysis, and widespread muscle atrophy beginning in the pelvic limbs as the dogs become nonambulatory (Awano et al, 2009; Matthews and de Lahunta, 1985). The paresis becomes more symmetric and progresses to flaccid tetraplegia in dogs with advanced disease. Widespread and severe loss of muscle mass occurs in the axial and appendicular musculature. Most previous reports attributed loss of muscle mass to disuse, but flaccidity in dogs with protracted disease suggests denervation. Cranial nerve signs include difficulty swallowing and inability to bark. Urinary and fecal continence usually are spared until the latter disease stage of LMN paraplegia. Electrodiagnostic testing is useful for detecting evidence of neuromuscular disease. Early in the progression of DM when UMN signs predominate, no spontaneous activity is detected by electromyography (EMG) and nerve conduction velocities are within normal limits. Later in the disease course with the emergence of LMN signs, EMG reveals multifocal spontaneous activity in the distal appendicular musculature. Fibrillation potentials and positive sharp waves are the more common waveforms recorded. Recordings of compound muscle action potentials (M waves) from stimulation of the tibial and ulnar nerves have been found to show temporal dispersion and decreases in amplitudes (Awano et al, 2009). The proximal and distal motor nerve conduction velocities have been found to be decreased when compared with the normal reference range. These findings provide evidence of motor axonopathy and demyelination in the late disease stage of DM. Because a variety of common acquired compressive spinal cord diseases can mimic DM by compromising the UMN pathways, a definitive diagnosis of DM can be accomplished only at postmortem examination. The pathologic features of DM include axonal degeneration with secondary demyelination and astroglial proliferation (sclerosis) in all spinal cord funiculi but consistently most severe in the dorsal portion of the lateral funiculus and in the dorsal columns of the middle to lower thoracic region (March et al, 2009). Absence of neuronal cell body degeneration or loss in the ventral horn of the spinal cord is not a prominent histopathologic finding. Histopathologic studies of tissue from dogs in late disease stage with LMN signs have documented denervation atrophy in muscle, nerve fiber loss with axonal degeneration, and secondary myelin loss in myelinated fibers of peripheral nerves (Awano et al, 2009; Shelton et al, 2012). Similarly, in ALS patients and DM-affected dogs, muscle biopsy specimens show evidence of reinnervation in the early disease stages (Shelton et al, 2012). Based on clinical signs and pathologic findings, DM is now considered a multisystem disease involving central and peripheral axons that has similarities to UMN-onset ALS.
Canine Degenerative Myelopathy
Pathophysiology
Clinical Spectrum
Early Disease (Upper Motor Neuron Signs)
Late Disease (Lower Motor Neuron Signs)
Diagnostic Approach
Neurodiagnostic Testing
Neuropathologic Features
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


