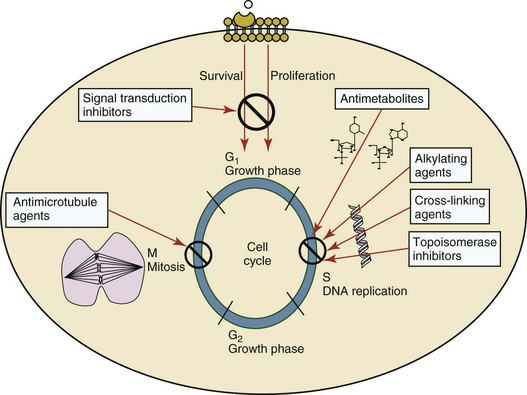
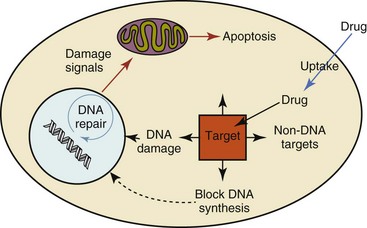
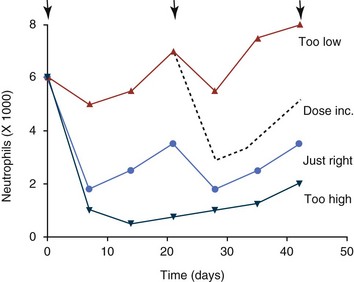
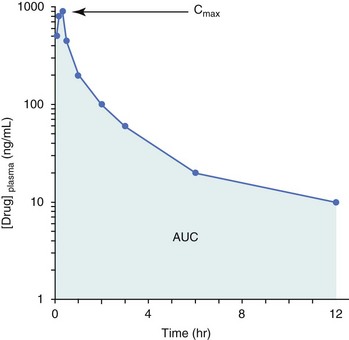
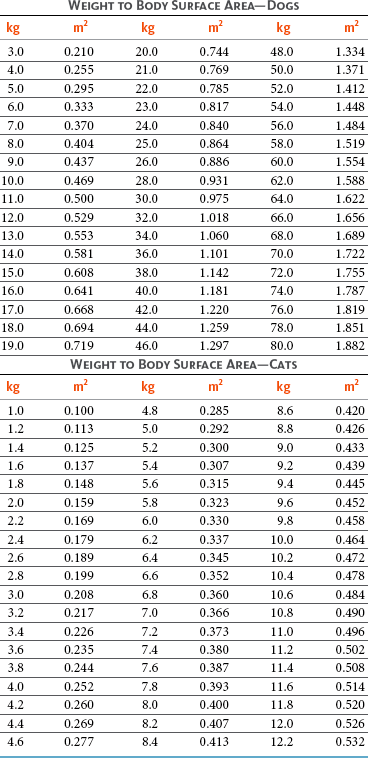
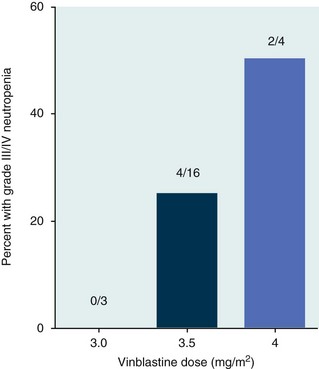
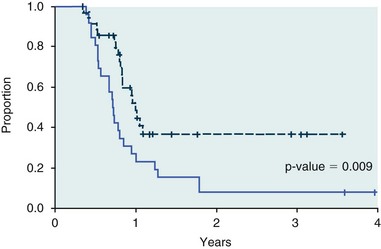
11 The use of chemical elixirs for the treatment of cancer can be traced through the medicinal customs and practices of a number of cultures.1 The modern use of pharmacologic agents to treat cancer began in the mid-1940s when Alfred Gilman and Louis Goodman showed the efficacy of nitrogen mustard in tumor-bearing mice, and these results were quickly translated and verified in human patients. These results and the efforts of others such as Sydney Farber with antifolates and George Hitchings and Gertrude Elion with purine analogs rapidly advanced the growing interest of treating cancer with drugs. The beginning of a systematic screening program for anticancer drugs at the National Cancer Institute (NCI) in 1955 set the framework for cancer chemotherapy development in both the public and private sectors and led to the characterization of many of the agents still in clinical use today.2,3 The basis of anticancer drug activity is the targeting of dividing cells through interference with processes involved in progression through the cell cycle. As shown in Figure 11-1, the major classes of drugs used to treat cancer work at various steps in the processes of DNA replication (S phase) and subsequent cell division (M phase). Another set of therapeutic agents, the signal transduction inhibitors, work by interfering with the signaling processes that trigger entry into the cell cycle and continuing cellular proliferation. This newer class of agents is discussed in Chapter 14, Section B, of this text. DNA synthesis is a complicated process involving anabolic processes to create the purine and pyrimidine nucleotide triphosphates required for replication, unwinding of the template DNA to provide access to the replication machinery, and the high fidelity process of creating complementary strands. Anticancer drugs work at all of these levels of DNA synthesis, including the antimetabolites that inhibit anabolic processes required for providing the nucleotide building blocks, topoisomerase inhibitors that interfere with the enzymatic process of DNA unwinding, cross-linking agents that through either interstrand or intrastrand interactions block the processes of strand separation and template processing, and the alkylating agents that interfere with the replication machinery through multiple mechanisms of altered binding and base recognition. The resulting effects of interacting at these levels of DNA replication can include the generation of DNA strand breaks, incomplete replication, and triggering of apoptotic signaling such that cell death is the ultimate result. As a preliminary metric, the clinical measurements of the tumor response to cancer chemotherapy are useful for predicting the impact of treatment on the extent of disease or time interval of tumor control. Table 11-1 describes conventional measures of treatment response. Table 11-1 Measures of Response in Cancer Therapy and Treatment Individual cell sensitivity to anticancer agents has been addressed empirically through the screening of tumor cell panels associated with a given histotype. The NCI60 human tumor cell line panel is the most well characterized and studied compilation with more than 100,000 compounds and 50,000 extracts from natural products screened to date.4 Further, rich gene expression and other characterizations of these cell lines exist in public databases so that drug sensitivity and genotypic characteristics can be considered.5 The use of canine tumor cell line panels to screen drug sensitivity is becoming established6 as a viable way to identify potential drug combinations for further testing as well. Chemosensitivity depends on a number of factors, including drug uptake into the cell, interaction with a cellular target, generation of lethal damage to important cellular macromolecules, repair of potentially lethal damage, and the cell’s response to generated damage as depicted in Figure 11-2. Uptake of some cancer chemotherapeutic agents occurs via passive diffusion due to their lipid-soluble properties; other compounds are actively transported into tumor cells. Melphalan is actively transported into cells by two amino acid transporters,7 and blocking transport with amino acid substrates or analogs can significantly reduce cytotoxicity.8 Other examples include nucleoside transporters used by Ara-C9 and gemcitabine10 and the reduced folate carrier system involved in methotrexate uptake.11 The intracellular target(s) for specific chemotherapeutic agents can play a role in determining sensitivity based on their levels and the nature of the interaction. For example, topoisomerase IIα levels can play a role in the sensitivity of tumor cells to doxorubicin12,13 as altered levels via decreased gene copy or transcriptional downregulation leads to a decrease in sensitivity (resistance). The opposite is true for thymidylate synthetase levels and 5-fluorouracil (5-FU) toxicity where increased levels of enzyme correlate to a decrease in sensitivity to 5-FU.14 Although the nature of the interaction with the target is different for doxorubicin and 5-FU, the fact that altered target levels can modulate response shows how quantitative interactions with the target can alter drug sensitivity. The extent of cellular damage, potential repair of that damage, and the cellular response occur in a tightly knit continuum that determines cellular fate. The generation of cellular damage is a consequence of interaction with a cellular target and can be either a primary or secondary event. In general for DNA-damaging agents, the resulting DNA lesions are caused by the interplay of DNA binding and DNA repair. For example, DNA strand breaks that result from O-6-methyl guanine lesions are due to aberrant mismatch repair processes and subsequent replication.15 DNA damage also triggers response pathways that can result in cell cycle arrest to allow for repair and subsequent survival or the triggering of apoptotic machinery that ultimately results in cell death. The definition of cellular response, whether mitotic catastrophe, apoptosis, necrosis, autophagy, or cellular stasis, depends on an intricate interplay of survival and death signaling and is often specific to the agent, dose at the critical target, and the cell lineage.16 Alterations in proapoptotic and antiapoptotic signaling clearly play a role in tumorigenesis and response to therapy,17 and the impact of antiapoptotic signaling in lymphoma by the mediators, bcl-2 and survivin, seem the most clear in regards to both chemosensitivity18–20 and response to therapy in humans and dogs.21–23 However, a clear understanding of the role of damage response and active cell death pathways in chemotherapeutic sensitivity and response in solid tumors is still lacking. Acquired resistance, or selection of resistant cells, during the treatment process is thought to be one of the major mechanisms of therapeutic failure during cancer drug therapy. Resistance of tumor cells to chemotherapeutic agents can depend on the drug and its mechanism or be through a multidrug mechanism. In general, the development of acquired resistance to a specific agent can come via a variety of mechanisms associated with drug uptake, drug metabolism/detoxification, target modification, damage repair, or damage recognition and response. Changes in cellular drug levels can come about due to either a decrease in drug uptake or through increased efflux. Decreased expression of transporters known to play a role in drug uptake have been observed in response to treatment with melphalan in human breast cancer cells24 and acquired resistance to methotrexate in KB cells,25 with the resulting cells showing drug resistance that correlated with the lower intracellular drug levels. The induction of drug efflux pumps in response to drug treatment is a primary mechanism for multidrug resistance and will be discussed later in this section. Alterations in metabolic or detoxification pathways within tumor cells are another mechanism by which acquired resistance occurs. Due to the fact that many chemotherapeutic agents are electrophilic based on their DNA-binding properties, enhancement of conjugation reactions with nucleophiles such as glutathione is a plausible mechanism of resistance.26 Induction of glutathione S-transferases has been shown to be a mechanism by which tumor cells can acquire resistance to nitrogen mustards.27,28 Although tumor cells themselves generally have limited drug metabolism capabilities, some metabolic pathways can play a role in the resistance phenotype. For example, the sensitivity of tumor cells to 5-FU is inversely correlated with the expression of dihydropyrimidine dehydrogenase,29–31 the enzyme predominantly responsible for the metabolism of 5-FU to the inactive 5-FUH2 metabolite.32 For prodrugs such as gemcitabine, which must be phosphorylated to the di- and tri-phosphate forms prior to eliciting an inhibitory effect on DNA synthesis,33,34 the enzyme responsible for this metabolic activation, deoxycytidine kinase,35,36 has been shown to be decreased in pancreatic tumor cells made resistant to this drug.37,38 This interplay between metabolic detoxification and activation, predominantly with drugs that are nucleotide analogs, leads to complex scenarios involving the upregulation of catabolic processes and the downregulation of anabolic processes regarding the cellular pharmacology of these cytotoxic agents. Modifications in the cellular target of a given drug usually pertain to mutations in the target protein leading to a decrease in affinity or absence of drug interaction. These modifications can include a decrease in the levels of a specific target responsible for the generation of a toxic product, increases in target levels to ameliorate the effect of target inhibition, or target mutations such that the drug can no longer interact in a manner detrimental to the tumor cell. Decreased topoisomerase II gene expression and activity has been observed in human lung and colon cells with acquired resistance to the epipodophyllotoxins,39 etoposide, and teniposide, whose antitumor activity involves topoisomerase II–dependent DNA strand break formation.40,41 Target amplification as a mechanism of acquired resistance has been observed in methotrexate resistance where gene amplification and increased dihydrofolate reductase (DHFR) levels allow for cells to overcome DHFR inhibition by this agent.42 Mutations in targets such as β-tubulin in the case of paclitaxel43 and topoisomerase I for camptothecin44 affect binding of drug and interaction with the target, thus generating tumor cells resistant to the toxic mechanism of these agents. These examples of altered target levels and structures show that drug resistance can come about via either quantitative or qualitative change in the nature of the interaction of drug and target and will depend on the impact of these changes on tumor cell growth both in the absence and presence of the selective agent. Damage repair in cancer cells treated with chemotherapy commonly refers to DNA repair processes since a majority of chemotherapy agents work at the level of the DNA. Resistance conferred through alteration in DNA repair include not only the induction of specific processes to repair discrete lesions but also more global DNA repair processes, such as postreplication and mismatch repair.45 Multiple studies have shown that enhanced removal of platinum adducts from tumor cell DNA correlate with acquired resistance46–48 to cisplatin, although the exact mechanism(s) and protein(s) responsible for repair of these lesions are unknown. The bulky DNA adducts generated by many cancer chemotherapeutic agents can cause replicative gaps in DNA that require postreplication surveillance and repair. The ability of cells to bypass these bulky lesions and interstrand cross-links during DNA replication has been found to be an important process in tolerance to agents causing these types of DNA damage (cisplatin, mitomycin C, melphalan); multiple DNA repair pathways can account for this release from DNA replication block.49 The fact that DNA repair pathways and processes are redundant and nondiscrete and that both lesion specific and global processes seem to play a role in determining drug resistance highlights the problems associated with attributing specific proteins or pathways to specific resistance phenotypes. Some mechanisms of acquired resistance result in a phenotype in which the tumor is resistant to multiple chemotherapeutic agents or multidrug resistant (MDR). Some of the mechanisms discussed previously, including DNA repair, enhanced metabolism, or detoxification, and resistance to apoptosis can result in resistance to multiple agents; however, the MDR phenotype generally refers to tumor cells expressing individual or multiple members of the adenosine triphosphate (ATP)-binding cassette (ABC) transporter family who play a primary role in active efflux of drugs from cells. Forty-eight ABC genes have been identified in the human genome,50 and currently, fifteen members of the ABC transporter family have been recognized that include a cancer chemotherapeutic as a substrate for transport.51 These include the well-studied and characterized PGP/MDR1 (ABCB1), MXR/BCRP (ABCG2), MRP1 (ABCC1), and MRP2 (ABCC2). The basic function of the ABC transporters is conserved across the family and involves the ATP-dependent transport of xenobiotics and endogenous substrates from the inside of the cell to the extracellular space. The role of ABC transporters in multidrug resistance of canine and feline cancers is poorly explored; however, ABCB1 is expressed in canine lymphoma,52 canine mammary tumors,53 and canine and feline primary pulmonary carcinomas.54 ABCC1, ABCC2, ABCC5, ABCC10, and ABCG2 have all been shown to be expressed in canine mammary tumors as well.53,55 The normal tissue distribution of the ABC transporters is also beginning to be investigated in dogs, with initial studies showing similar tissue distributions and presumed function, although there appears to be some partial differences in relative expression in various tissues.56,57 A recent study has shown that feline ABCG2 has specific amino acid changes that lead to transporter dysfunction with regard to a number of substrates, suggesting that cats may have altered pharmacokinetic disposition for drugs that are ABCG2 substrates.58 The success of combination chemotherapy as compared to single-agent treatment is attributed to the overcoming of both natural and acquired resistance of tumor cells, as well as use of agents that differ in dose-limiting side effects. A premise of cancer chemotherapy is to kill the largest fraction of tumor cells possible with each dose, with the dose and timing of each therapy based on the normal tissue tolerance. Therefore a strategy that allows for more intensive cycles of fractional cell killing without exacerbating the recovery of normal tissue damage is preferred. Combination chemotherapy has been shown to be curative in humans with acute lymphocytic leukemia (ALL), Hodgkin’s disease, histiocytic lymphoma, and testicular carcinoma, whereas single-agent therapy was not.59 The success of combination therapies as opposed to single-agent therapy is best illustrated in veterinary oncology by treatment protocols for canine lymphoma. Doxorubicin is the most active single-agent therapy tested against canine lymphoma; other combination protocols that generally consist of cyclophosphamide, vincristine, and prednisone result in similar outcomes (Table 11-2). However, these same combination protocols with doxorubicin included empirically seem to increase both the median remission and median survival times over either doxorubicin alone or a combination protocol that excludes doxorubicin. It should be noted that the populations represented in Table 11-2 may not be homogeneous, and this data does not represent a formal reanalysis of these data sets. Table 11-2 Response of Canine Lymphoma to Single-Agent Doxorubicin, Combination Protocols, and Combination Protocols Including Doxorubicin* *Values represent the mean ± standard deviation (SD) from the cited studies. The combination protocols used included cyclophosphamide, vincristine, and prednisone, with methotrexate and actinomycin D included in some. †Number represents the total number of dogs, with the number of individual studies in parentheses. ‡Data for doxorubicin alone studies were from references 60–64. §Data for combination studies were from references 61, 65–68. Dosing conventions have been developed from formal phase I studies for an increasing number of agents investigated specifically in companion animals. Nonetheless, suggested starting doses represent an estimate of the MTD from a small population of animals and safe individual patient dosing may vary substantially. There are numerous reasons for pharmacokinetic variability in cancer chemotherapy among a population of patients.77 Concurrent illness or organ dysfunction, extreme tumor burden, specific breed sensitivities (e.g., Collie with ABCB1 mut/mut) or idiosyncratic considerations (anticipated drug-drug interactions or drug allergies) will mandate modification of the protocol and dosing. Concurrent illness and organ dysfunction can also have profound effects on selection of anticancer agents and dosing. In general, predictable dose adjustments for pets with renal or hepatic disease have not been developed and treatment should be approached conservatively. Interestingly, in cats, the glomerular filtration rate (GFR) can be used to define an individual dose for carboplatin that will permit some patients with renal disease to be safely dosed that would not have been safe if dosed by conventional methods.78 Dose adjustments of 30% to 40% have been recommended for drugs that are ABCB1 substrates in Collie-type breeds in which an ABCB1 (mut/mut) phenotype is confirmed, and even in dogs with an ABCB1 (wt/mut) phenotype, dose adjustments may need to be made.79 Chemotherapeutic dosing in obese patients often raises questions about drug partitioning in lipid storage sites around the body. Distribution of many pharmaceutical agents may be affected in obese patients; however, there is no accepted scale for empiric dose adjustments in humans. Individual factors such as the specific drug, degree of obesity, and other comorbidities may convince a clinician to dose reduce or cap the dose of a chemotherapeutic agent.80 Some reviews suggest that dose reductions based on body mass may ultimately be detrimental to outcomes in obese patients.81 It is the initial chemotherapeutic intervention that is expected to result in the greatest opportunity to benefit the patient; therefore taking the time to assess the patient’s specific medical limitations and then proceeding with thoughtfully designing, administering, and completing a therapeutically robust protocol are highly desirable. As individual patient tolerance and response to each compound in a multiagent protocol is observed, future modifications may be anticipated more accurately. The greatest benefit achievable with anticancer cytotoxic therapy requires a commitment to dose intensity. Optimal dose intensity demands therapeutic monitoring in order to either reduce or increase the dose based on the patient’s capacity to maintain a high quality lifestyle during effective therapy. The decision to increase the dose of an agent is conceptually challenging but important. In order to make a recommendation to increase dosing of a cytotoxic compound, owner understanding and monitoring of the patient’s white blood cell values and clinical events during the first treatment cycle are critical. A dose of a cytotoxic agent that does not result in any change in the target normal tissue (e.g., blood neutrophil count) is likely ineffective and could be increased 10% at the next infusion with continued follow-up to determine adequacy of dose adjustments (Figure 11-3). Dose reductions are deleterious to the optimum delivery of chemotherapy but are to be anticipated. Specific guidelines for dose adjustments of antineoplastic agents are not standardized. In general, a 20% to 25% reduction is recommended for the subsequent dose for patients experiencing a moderate or severe dose-limiting toxicity, such as neutropenia or emesis. Close monitoring and preemptive handling of signs may permit successful management of some potential future clinical signs, and clinical decisions are based on the extent and severity of the resulting signs as described in Table 11-3. Table 11-3 Guidelines for Common Chemotherapy-Induced Toxicity SQ, Subcutaneous; IV, intravenous; CBC, complete blood count. *Fluoroquinolone (enrofloxacin 5-10 mg/kg once daily). †Ampicillin 20 mg/kg oral (PO) or IV three times/day (TID) ± cephalosporin 20 mg/kg oral or IV TID. ‡Maropitant 2 mg/kg PO or SQ once daily × 3-5 days—dogs; 1 mg/kg PO or SQ once daily × 3-5 days—cats (not labeled for cats). §Famotidine 0.5-1.0 mg/kg PO, SQ, or IV. ¶Loperamide 0.08 mg/kg PO TID; tylosin 10 mg/kg PO TID; metronidazole 15-25 mg/kg PO BID. Delayed acute effects from chemotherapy often include bone marrow suppression and nausea, vomiting, and diarrhea. In the majority of instances, these effects are self limiting and the incidence of hospitalization for such problems is low. Table 11-3 reviews the general therapeutic strategies for management of the most common types of adverse events experienced in companion animals following chemotherapy.82 Examples of potential cumulative and/or chronic toxicity include hepatic dysfunction after multiple doses of cyclohexylchloroethylnitrosourea (CCNU), cardiac abnormalities after exceeding a safe cumulative dose of doxorubicin, and renal disease after cisplatin use in dogs or doxorubicin use in cats. Screening recommendations and strategies to reduce the risks of such chronic effects have been developed and are incorporated into standard protocol procedures. It is critical to the success of treatment that owners be thoroughly informed about monitoring guidelines for the general signs and symptoms of chemotherapy-induced toxicity. Online educational resources for owners are readily available at www.csuanimalcancercenter.org. It is advisable to instruct the owner regarding monitoring and early responses when their pet experiences nausea and vomiting, diarrhea, or hematuria and it is important to inform the owner about how to obtain an accurate body temperature. These “at home” aids will allow the clinician to assess the management options should a concern arise. In general, safety concerns for cancer therapy are only applied to the patient with regard to the impact of drug treatment. An issue with cytotoxic chemotherapy, however, is the preparation and distribution of the drugs, as well as active drug eliminated in the urine and feces of the patient and the potential exposure of health professionals, caregivers, and others in the home. The preparation of these drugs should be done under strict regulations and involves the use of protective clothing, gloves, masks, and chemical hoods. Studies have characterized the potential impact of secondary exposure to these agents on oncology health workers in human medicine with regard to cancer prevalence, reproductive risks, and acute toxicities, and the results show little risk.83 However, these results are a reflection of exposure in trained cohorts of individuals working in human medicine where fecal and urinary exposures are more limited. Clients with animals undergoing cancer therapy need to be informed of potential risks and safety precautions that need to be adhered to when dealing with oral medications and pet excrement. Simple precautions such as wearing gloves when handling oral medications and not opening capsules or splitting tablets are essential. Oral suspensions of these agents should be avoided. Urinary levels of some active drugs may remain high for days after treatment84 and fecal excretion may also be expected. Therefore careful avoidance, collection, and disposal of urine and feces must be recommended, as well as having pregnant women, small children, and immunosuppressed individuals in particular avoid any contact with pet wastes for a defined time period following treatments. Preparation of guidelines for minimizing exposure to individuals and the environment should be prepared and distributed to clients for general, as well as drug-specific, instructions. Pharmacokinetic (PK) considerations in cancer drug therapy are important due to the relationship between drug exposure and pharmacodynamic (PD) response, whether efficacy or toxicity, that is more exact than the relationship between drug dose and PD response.85 PK considerations are also important with regard to interactions with other drugs,86 herbal products,87,88 and genetic differences among breeds and individuals89 that can cause changes in drug exposure at a given dose. Cytotoxic chemotherapy is usually dosed on an MTD-based schedule reflecting only acceptable toxicity and thus limits any informative role of drug half-life and effective therapeutic concentrations from initial dosing considerations. The most important PK parameters are those that have a relationship with either a response to therapy (efficacy) or toxicity, which is most often either the area under the plasma/serum concentration versus time curve (AUC) or the maximum drug concentration (Cmax) achieved, illustrated in Figure 11-4. The relationships of AUC and Cmax in the clinical pharmacology of doxorubicin illustrate the complex associations with PK considerations. The Cmax during doxorubicin infusion in humans is related to the incidence of cardiotoxicity both in adult90 and pediatric91 patients but is also associated with longer remissions in leukemia patients.92 A relationship between AUC values and decreased white blood cells has also been established with doxorubicin.93 However, no clear relationships between AUC and efficacy exist.94 These data have allowed for adjustments in doxorubicin dosing protocols so that intermediate infusion times (10 to 30 minutes) are utilized to decrease the Cmax and thus cardiotoxicity while still maintaining peak levels associated with effective therapy. PK studies that relate drug exposure to responses are an important first step in establishing relationships that may be exploited for dose modification based on patient characteristics or therapeutic drug monitoring. These data are generally lacking for drugs used to treat cancer in companion animals with a few exceptions. Studies on the PK and myelotoxicity of carboplatin in cats have shown a clear relationship between drug exposure and the neutrophil nadir as well as drug clearance and GFR (Figure 11-5). The fact that PK parameters can be correlated both with a toxic endpoint and a physiologic function allows for the calculation of a dosing metric relating the GFR of an individual cat to a dose that produces a drug exposure (AUC) that results in acceptable toxicity.78 It remains to be determined whether such individualized dosing results in improved outcome in a heterogeneous population. Current drug-dosing convention for cancer chemotherapeutic agents is the use of body surface area (BSA) for dose normalization (mg/m2). Exceptions to this paradigm are the use of body weight (mg/kg) for dogs that weigh less than 15 kg and for cats with doxorubicin dosing based on empiric evidence showing a better toxicity profile for smaller dogs when mg/kg dosing is used.95 The approximate calculation for BSA in dogs and cats based on weight is as follows: Figure 11-5 Relationship between (A) neutrophil nadir and carboplatin exposure and (B) platinum clearance and glomerular filtration rate (GFR) in cats being treated for cancer. Rights were not granted to include this figure in electronic media. Please refer to the printed book. (From Bailey DB, Rassnick KM, Erb HN, et al: Effect of glomerular filtration rate on clearance and myelotoxicity of carboplatin in cats with tumors, Am J Vet Res 65:1502, 2004.) Am J Vet Res where A is equal to 10.1 for dogs and 10.0 for cats. The implementation of this equation relating body weight in kilograms to BSA in meters squared is shown for dogs and cats in Table 11-4. PD considerations for cytotoxic chemotherapy are generally related to standard measures of response (i.e., complete remission [CR], partial remission [PR], stable disease [SD]) and toxicity.96 A majority of the literature in veterinary oncology relates PD responses to specific drugs or combinations, doses, or schedules. Figure 11-6 shows the relationships between vinblastine doses and the incidence of grade III or IV neutropenia observed in a phase I cohort of dogs.97 These results relate a dose to a PD response with the absence of exposure PK data. PD endpoints can also be used as indicators of efficacy and potentially as targets of therapy. The proportion of dogs in remission following treatment for lymphoma is increased in the subgroup experiencing grade III or IV neutropenia compared to the group that did not show that level of observed toxicity (Figure 11-7).98 In this example, the therapeutic response was related to overall drug effects on normal tissues as indicated by the degree of neutropenia (PD response), whereas DI did not show a significant difference. Again, these data did not include exposure PK assessment and in this case only relate the therapeutic outcome to an observed drug response. A lack of complete PK/PD data relationships in veterinary medicine reduces the opportunities for therapeutic drug monitoring and the potential for optimizing efficacy. Pharmaceutics is the science associated with dosage form design with regard to formulation and optimizing drug delivery via a specific route. For example, improved formulations of clinical agents such as paclitaxel have made these compounds available for use in veterinary patients. The excipient (drug carrier) used in the original clinical formulation of paclitaxel (Taxol) was Cremophor EL, which causes histamine release and unacceptable toxicity when used in dogs and cats.99,100 A new water-soluble formulation of paclitaxel (Paccal Vet) has been tested and shown to be effective without associated hypersensitivity reactions in dogs with mast cell tumors. The ability of new formulations and delivery methods to alter the efficacy and toxicity profile of agents is a rapidly expanding field. It is expected that new technologies in drug formulation and targeting will be incorporated into veterinary medicine to alter drug delivery and distribution in a more favorable manner. Basic Pharmacology: Mechlorethamine is frequently referred to as “nitrogen mustard” and was the first cytotoxic agent to show antineoplastic activity.101–103 Mechlorethamine undergoes spontaneous hydrolysis to 2-hydroxyethyl-2-chloroethylmethylamine and bis-2-hydroxyethylmethylamine, yielding nucleophilic reactive centers capable of forming DNA cross-links.104 Clinical Pharmacology: Mechlorethamine rapidly disappears from the plasma following intravenous (IV) administration primarily through spontaneous degradation, although some percentage of the drug is enzymatically metabolized.105 Mechlorethamine uptake into cells seems to be carrier mediated with decreased uptake as a mechanism for resistance.106 Clinical Use: Mechlorethamine is used predominantly in multiagent protocols for lymphoma in dogs.107–109 Experience with mechlorethamine as a single agent is not reported, although gastrointestinal (GI) and bone marrow toxicity are dose-limiting toxicities of conventional mustargen, vincristine, prednisone, and procarbazine (MOPP) protocols. Dosing of mechlorethamine in these protocols is reported as 3 mg/m2 IV on days 0 and 7 of a 21-day cycle. Basic Pharmacology: Melphalan (l-phenylalanine mustard) is a nitrogen mustard containing DNA cross-linking agent with a similar structure and pharmacology to chlorambucil. The major difference is that melphalan is actively transported into tumor cells by amino acid transporters7 and its uptake can be blocked by the amino acid leucine. Melphalan has direct alkylating activity and does not require metabolic activation. Clinical Pharmacology: Melphalan can be given orally with an oral bioavailability of approximately 30%. A relatively high percentage of melphalan (20% to 35%) is excreted unchanged in the urine with a majority of the remainder of the dose undergoing spontaneous chemical decomposition to inert products.110 The primary toxicity is myelosuppression. Clinical Use: The primary indication for melphalan in companion animals is for management of myeloma. The initial dose of 0.1 mg/kg PO daily for 10 to 14 days should be reduced to 0.05 mg/kg PO daily based on control of the paraproteinemia and hematologic screening for both dogs and cats. Alternate dosing regimens have also been used for dogs: 7 mg/m2 PO daily for 5 days every 3 weeks or 2 mg/m2 PO daily for 10 days with a 10-days-off cycle and repeated as needed. Basic Pharmacology: Cyclophosphamide (CP) is a nitrogen mustard–containing prodrug that is inactive in the absence of metabolic activation that occurs via microsomal mixed function oxidases predominantly in the liver.111 The activation of CP involves ring oxidation to 4-hydroxycyclophosphamide (4-OHCP), spontaneous and reversible ring opening to the amino aldehyde aldophosphamide, and the subsequent irreversible breakdown of aldophosphamide to phosphoramide mustard and acrolein. Phosphoramide mustard is considered the most active CP metabolite and is capable of bifunctional alkylation and cross-link production.112 Clinical Pharmacology: Recent studies have characterized the PK of CP and the 4-OHCP metabolite following both IV and oral dosing in dogs.113 The results of this study show that although exposure to CP is decreased following oral dosing, the overall exposure of 4-OHCP is similar when CP is dosed either intravenously or orally. The major dose-limiting side effects of CP are neutropenia and thrombocytopenia. GI toxicity (nausea and vomiting) is not common in dogs but has been observed in cats.114 Other common toxicities include alopecia in dog breeds with continually growing hair and hemorrhagic cystitis. Although hemorrhagic cystitis is uncommon at conventional doses, furosemide is often administered (1 mg/kg subcutaneous [SQ] or IV) prior to IV injection and precautions are taken at home to encourage vigorous hydration and frequent urination. CP should be discontinued permanently if hemorrhagic cystitis occurs and chlorambucil may be substituted in a multiagent protocol. Urine culture, antiinflammatory drugs, and antispasmodics should be initiated at the onset of clinical signs of cystitis. Aggressive intravesical therapy or even surgery may be necessary in severe instances. Clinical Use: CP is commonly included in multiagent protocols for lymphoma in both dogs and cats. It is effectively administered as a bolus dose (250 mg/m2) by either an oral (PO) or IV route in the dog.113 A fractionated dosing schedule is also used in some protocols (50 mg/m2 for 3 to 4 consecutive days after doxorubicin).115 Metronomic therapy, the use of very low dosing for prolonged periods, has also been developed for CP. The rationale and application of metronomic therapy is covered extensively in Chapter 14, Section C, of this text. CP is administered IV (200 to 250 mg/m2) in many multiagent protocols for lymphoma in cats. The efficacy of oral dosing of CP has not been as carefully investigated in cats compared to dogs although it may be safely administered at doses of 300 mg/m2 PO every 3 weeks in multiagent protocols.116 Basic Pharmacology: Ifosfamide (IP) is a nitrogen mustard–containing prodrug that like CP requires metabolic activation by microsomal mixed function oxidases prior to generating the isofosforamide mustard metabolite capable of bifunctional alkylation.117 Clinical Pharmacology: The major difference between the clinical use of IP and CP is due to differences in the relative metabolism of the parent drugs, with dechloroethylation accounting for up to 25% of the metabolism of IP,118 whereas this number is much smaller for CP. This difference in metabolism accounts for an increase in the formation of the neurotoxic metabolite chloroacetaldehyde following IP dosing and potentially for the less favorable metabolism profile observed with IP following oral dosing.119 The primary toxicity associated with IP treatment is a dose-related myelosuppression, but nephrotoxicity and damage to the bladder epithelium are not uncommon. Vigorous hydration is required with IP administration; in addition, mesna, a urinary epithelial protectant, must be administered to avoid severe cystitis. Clinical Use: Ifosfamide has been evaluated in dogs and cats with cancer and is primarily recommended for management of sarcomas. The recommended dose for dogs is 300 to 350 mg/m2 IV slow infusion with diuresis every 3 weeks, and for cats, the recommended dose is 900 mg/m2 IV slow infusion with diuresis every 3 weeks.120,121 The basis for such discrepancies in the MTD between species is not understood but reflects profound and interesting differences in metabolism pathways and most likely reduced generation of bioactive metabolites. A phase II study in feline vaccine sarcomas reported moderate objective response rates.122 Basic Pharmacology: Chlorambucil (p-bis[chloro-2-ethyl] amino-phenyl-4-butanoic acid) is a nitrogen mustard derivative that enters cells via passive diffusion123 and has direct bifunctional alkylating ability124 that is responsible for the cytotoxic activity. Clinical Pharmacology: Chlorambucil is orally bioavailable with rapid absorption. Hepatic metabolism is extensive with the pharmacologically active phenylacetic acid being the primary metabolite and is presumably responsible for much of the clinical activity.125,126 The major dose-limiting toxicity is myelosuppression, including neutropenia and thrombocytopenia.
Cancer Chemotherapy
General Principles of Cancer Chemotherapy
Indications and Goals of Therapy
Response Term
Abbreviation
Description
Complete remission/response
CR
Complete disappearance of tumor(s) and symptoms of disease.
Partial remission/response
PR
Decrease in tumor volume of ≥50% or decrease in tumor maximum diameter of >30%.
Stable disease
SD
Neither an increase nor a decrease in tumor size or disease symptoms (e.g., ±20% diameter changes).
Progressive disease
PD
Increase in tumor volume of >25% or increase of tumor maximum diameter of >20%; appearance of new lesions.
Median duration of response/median duration of survival
MDR/MDS
The median value for a group of individuals treated with a given therapy in terms of the length of time they achieved a complete or partial remission (MDR) or length of survival following implementation of therapy (MDS).
Progression-free interval/progression-free survival
PFI/PFS
The amount of time elapsed without evidence of progressive tumor growth (PFI) or survival without progressive growth of the tumor from treatment start (PFS).
Disease-free interval/disease-free survival
DFI/DFS
The amount of time that elapses without disease recurrence (DFI) or survival (DFS) of the patient following therapy.
Tumor Susceptibility and Resistance
Tumor Cell Resistance
Combination Therapies
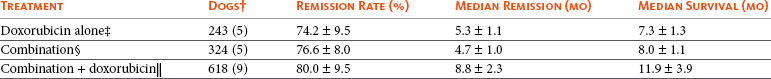
 Data for combination studies, including doxorubicin, were from references 68–76.
Data for combination studies, including doxorubicin, were from references 68–76.
Toxicities Associated with Drug Therapy of Cancer
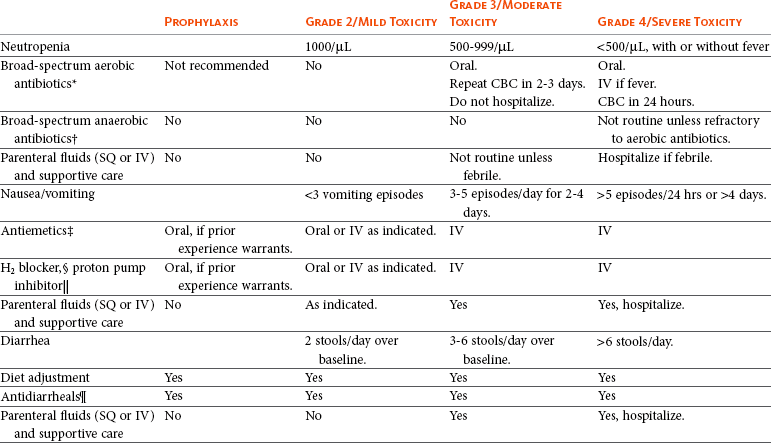
 Pantoprazole 1 mg/kg IV or SQ as needed.
Pantoprazole 1 mg/kg IV or SQ as needed.
Safety Concerns of Cancer Drug Therapy
Pharmacologic Principles in Cancer Therapy

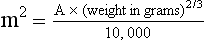
Pharmacodynamics
Pharmaceutics
Specific Chemotherapeutic Agents
Nitrogen Mustards
Mechlorethamine
Melphalan
Cyclophosphamide
Ifosfamide
Chlorambucil
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree