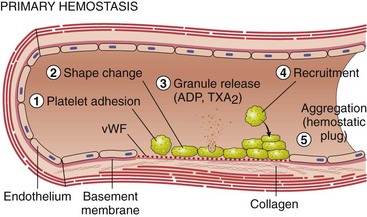
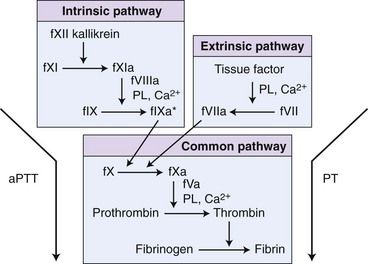
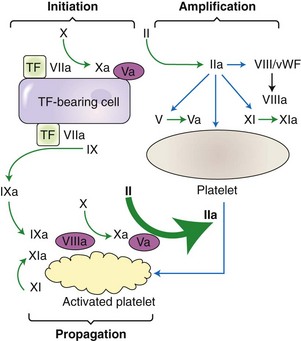
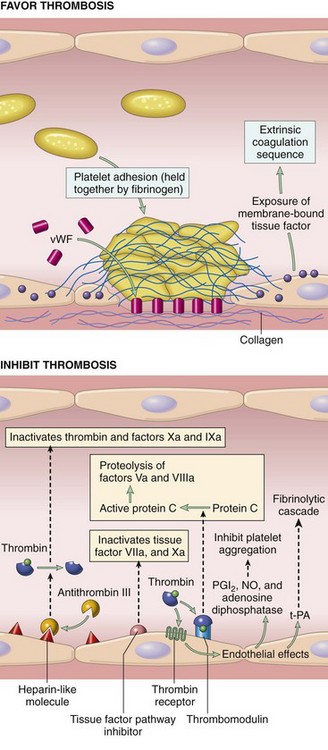
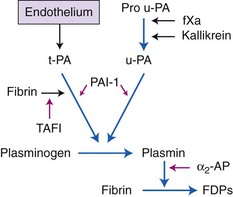
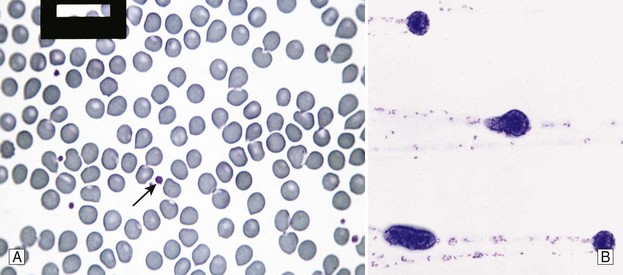
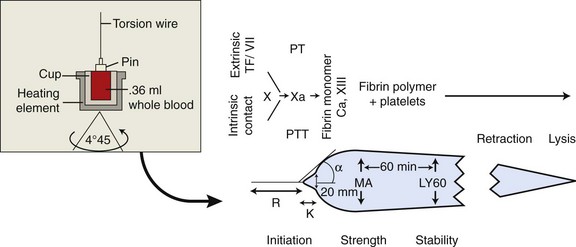
Chapter 7 Hemostasis is the process that maintains the integrity of a closed, high-pressure circulatory system following vascular damage.122 Even under the best of circumstances, surgery represents a major hemostatic challenge. This challenge is magnified in patients with inherited or acquired disorders that affect the hemostatic system. In human patients, approximately 50% of surgical complications are attributed to coagulation abnormalities, either hemorrhage or thrombosis, in the operative or postoperative period.206 Although the prevalence in small animals is unclear, there is little doubt that coagulation abnormalities significantly affect surgical morbidity and mortality. Successful surgical outcome demands not only technical expertise but the identification and management of disorders that compromise hemostasis or promote excessive thrombosis. This chapter aims to provide the surgeon or other practitioner with a solid basis from which to engage, rather than replace, the criticalist or internist. Early consultation and collaboration may be prudent and clinically useful. A list of abbreviations used in this chapter is provided in Table 7-1. Table • 7-1 Abbreviations Used in This Chapter Platelets are derived and released from progenitor megakaryocytes in the bone marrow at a rate of approximately 1011 platelets per day.278 They circulate as anucleate cells, with a life span of 6 to 8 days in the dog and cat, and provide a source of preformed chemokines that are stored in intracellular storage granules. In addition, activated platelets synthesize prostanoids, notably thromboxane A2 (TxA2), from arachidonic acid. This reaction is mediated primarily by the enzyme, cyclooxygenase (COX) -1. Following endothelial disruption, platelets adhere to subendothelial collagen via the platelet glycoprotein VI receptor, or to collagen-bound von Willebrand factor (vWF) via the glycoprotein Ib receptor (Figure 7-1).122 Adherence triggers a cascade of cytosolic signaling that results in stimulation of arachidonic acid metabolism and the release of granular contents (activation). Thrombin, generated by secondary hemostasis, is also a powerful platelet agonist. Activated platelets release secondary agonists, notably TxA2 and adenosine diphosphate (ADP), that recruit and activate additional platelets, thus amplifying and sustaining the initial response.93,122 The final common pathway for all agonists is the activation of the platelet integrin αIIbβ3 (formerly known as glycoprotein IIbIIIa) receptor.93,122 Agonist binding induces a conformational change in the receptor, exposing binding domains for fibrinogen. Binding results in interplatelet cohesion and aggregation. The Cascade Model of Coagulation The traditional model of coagulation consisted of a cascade of enzymatic reactions in which enzymes cleaved substrates to generate the next enzyme in the cascade (Figure 7-2).224 This model was divided into two pathways: the “extrinsic” pathway, initiated by tissue factor, and the “intrinsic” pathway, initiated through contact activation of fXII. Either pathway could activate fX to fXa, which (together with cofactor fVa) activated prothrombin (fII) to thrombin (fIIa), which then cleaved fibrinogen to form fibrin. This latter portion was referred to as the “common pathway.” Although separation of coagulation events into distinct pathways is valid for interpretation of in vitro coagulation testing, it has become evident that this model does not adequately explain coagulation in vivo.122,156 The substantial role of cellular components is not addressed by the cascade model. This model suggests independent and redundant pathways, while clinical manifestations of coagulation defects clearly contradict this concept. For example, although deficiencies of fXII cause marked coagulation test prolongation, they do not result in a bleeding tendency. In contrast, isolated deficiencies of the intrinsic pathway, such as hemophilia, result in profound bleeding in spite of an intact extrinsic pathway. A cell-based model of coagulation more accurately reflects coagulation in vivo.122,156,301 This model includes two fundamental paradigm shifts: (1) that tissue factor is the primary physiologic initiator of coagulation (contact activation playing no role in vivo), and (2) that coagulation is localized to, and controlled by, cellular surfaces.122,156 Coagulation occurs in three overlapping phases: initiation (on tissue factor–bearing cells), amplification or priming, and propagation (on platelets) (Figure 7-3).156,301 The initiation phase is the tissue factor–initiated (extrinsic) pathway that generates small amounts of thrombin. Tissue factor is a membrane protein, expressed on fibroblasts and other extravascular cells under physiologic conditions.234 Coagulation is initiated when vascular damage allows contact between plasma and tissue factor–bearing cells. Plasma fVII binds to tissue factor and is activated. The fVIIa-TF complex activates fX. The fXa that remains on the cell surface combines with fVa to produce small amounts of thrombin. The fVIIa-TF complex also activates fIX, which diffuses off the cell. The initiating and propagating steps of coagulation are sequestered to different cell surfaces.156 Platelets do not express tissue factor; coagulation can proceed only when extravascular tissue factor is brought into close proximity with platelets and coagulation factors. Moreover, platelets that are not activated do not present the procoagulant membrane that is essential for secondary hemostasis to proceed effectively.156 In the inactive resting state, neutral phospholipids are located on the outer surface of the platelet cell membrane, and the negatively charged phosphatidylserine is localized to the inner surface. When platelets are activated, they actively shuffle phosphatidylserine to the outer membrane surface. The expression of phosphatidylserine markedly increases the speed of coagulation reactions. Under physiologic conditions, cells outside the injured area do not express a procoagulant membrane. Consequently, thrombin generation is extremely slow and does not generate sufficient fibrin to form a clot. The normal endothelium controls platelet reactivity through three known inhibitors: prostacyclin (PGI2), ectoadenosine diphosphatase (ecto-APDase), and nitric oxide.93,174 Endothelial cells convert arachidonic acid to PGI2. (In contrast to platelet prostanoid synthesis, COX-2 is predominant in endothelial cells.73) Prostacyclin limits the platelet response to TxA2.93 Ecto-ADPase, an integral component of the endothelial surface, is substrate activated. The enzyme metabolizes ADP released from activated platelets, thus removing a major agonist and abrogating platelet activation and recruitment.93,230 Nitric oxide is constitutively produced by endothelial cells. It diffuses into platelets and decreases intracellular Ca2+ flux, thus suppressing the conformational change in the integrin αIIbβ3 receptor, and reducing the number and affinity of fibrinogen binding sites.93 Three natural anticoagulant pathways are described: antithrombin (AT), activated protein C, and tissue factor pathway inhibitor (Figure 7-4). AT (previously named ATIII) is a circulating α2-globulin, produced in the liver. It inactivates coagulation proteins that escape into the circulation from a site of injury. AT exerts its most significant anticoagulant effect by binding and inactivating thrombin and fXa.25,30 It also neutralizes other factors (IXa, XIa, XIIa) and kallikrein.33 The rate of neutralization is dramatically increased when AT is bound to heparan sulfates in the endothelium. In addition to its anticoagulant functions, AT inhibits neutrophil adherence and exerts potent antiinflammatory effects.182,271,370 Free thrombin that is not bound to AT binds to the endothelial surface receptor, thrombomodulin. The thrombin-thrombomodulin complex activates protein C.108,256 This reaction is augmented when protein C is bound to the endothelial protein C receptor. Activated protein C and its cofactor, protein S, inactivate cofactors fVa and fVIIIa, and this slows the rate of thrombin formation. Activated protein C also enhances fibrinolysis via the inactivation of plasminogen activator inhibitor-1 (PAI-1).33 Over and above its role in hemostasis, the activated protein C pathway plays a significant role in limiting inflammatory responses and decreasing endothelial cell apoptosis in response to inflammatory cytokines and ischemia.106,108,256 A third anticoagulant, tissue factor pathway inhibitor, synthesized and expressed by endothelial cells, regulates the early phases of coagulation. Tissue factor pathway inhibitor inhibits tissue factor and abrogates the initiation complex of factor VIIa-TF, as well as factor Xa.233 Antiangiogenic and antimetastatic properties are also described.9 Fibrinolysis is the enzymatic dissolution of fibrin. Plasminogen activators proteolytically convert the proenzyme, plasminogen, to plasmin (Figure 7-5).80,382 Plasmin degrades fibrin into soluble degradation products (fibrin split products or fibrin degradation products). Two plasminogen activators are described: tissue-type plasminogen activator (t-PA), and urokinase-type plasminogen activator (u-PA).80,99,294,382 t-PA is synthesized and secreted by endothelial cells. In an autoregulatory manner, fibrin acts as both cofactor for plasminogen activation and substrate for plasmin. In the presence of fibrin, the catalytic efficacy of t-PA increases 1000-fold.99,294 Because it is a necessary cofactor in the reaction, the degradation of fibrin limits further activation of plasminogen. u-PA is released as an inactive glycoprotein; hydrolysis by plasmin or kallikrein yields active u-PA.99,294 Fibrinolysis is controlled predominantly by PAI-1, α2-antiplasmin, and thrombin activatable fibrinolysis inhibitor. Of these, PAI-1 appears to be most important.99,382 It is primarily stored in platelet α-granules and is released upon platelet activation. PAI-1 inhibits both tPA and uPA.382 α2-antiplasmin, synthesized in the liver, inhibits plasmin.294 Endothelial thrombomodulin plays an important role in controlling fibrinolysis. In addition to activating protein C, the thrombin-thrombomodulin complex catalyzes the activation of thrombin activatable fibrinolysis inhibitor, which downregulates the cofactor activity of fibrin in plasminogen activation, thereby suppressing fibrinolysis.263 The pathway defined by thrombin, thrombomodulin, activated protein C, and thrombin activatable fibrinolysis inhibitor, therefore, creates a direct molecular connection between coagulation and fibrinolysis, such that the activation of one suppresses the activity of the latter. This pathway likely plays a key role in the balance between fibrin deposition and removal. Laboratory testing is essential for the identification and characterization of hemostatic defects. It is important to be aware, however, that in vitro tests do not accurately reflect in vivo hemostasis. Moreover, hemostatic testing makes high demands on sampling procedure; improper technique leads to artifactual results.12 Tests should always be performed and interpreted carefully, with their limitations in mind. Routine and screening hemostatic tests are presented here. The platelet count detects quantitative platelet disorders (thrombocytopenia). Enumeration is performed via automated cell counter or manually (by hemocytometer). Pseudothrombocytopenia is a common artifact that occurs when platelets in blood are not counted, resulting in falsely low counts. This usually results from platelet aggregation that occurs during sample collection. It is especially common in cats, reported in 71% of feline blood samples.268 Pseudothrombocytopenia is also frequent when platelet counts are obtained in cats via automated counters, because of the considerable overlap between erythrocyte and platelet volumes in this species.193 A similar artifact occurs in both dogs and cats when large platelets are present. For these reasons, low platelet counts must always be confirmed by blood smear examination. Examination of a blood smear allows for rapid estimation of platelet numbers (Figure 7-6). The feathered edge of the smear should be evaluated for platelet clumps that indicate pseudothrombocytopenia and the need for repeat sampling. The use of an ethylenediaminetetraacetic acid (EDTA)-rinsed syringe for venipuncture may help to reduce clumping. If clumping is not present, the platelet count can be estimated. This is achieved by multiplying the average number of platelets per high power field (within the monolayer of the blood film) by 15,000.320 The bleeding time is the duration of hemorrhage resulting from the infliction of a small standardized injury involving only microscopic vessels. The buccal mucosal bleeding time is the only reliable and reproducible method in small animals.172 Sedation is generally not required, except in cats and nervous dogs. The patient is restrained in lateral recumbency, and a strip of gauze, sufficiently tight to cause moderate mucosal engorgement, is tied around the maxilla to fold up the upper lip. A two-blade, spring-loaded device (Simplate II, Organon Teknika Corporation, Durham, NC) is used to make two 1-mm deep incisions in the mucosa of the upper lip. The incisions should be made at a site devoid of visible vessels and inclined so that the blood flows toward the mouth. Shed blood is blotted carefully with filter paper, taking extreme care not to disturb the incisions. The buccal mucosal bleeding time is the time from incision to cessation of bleeding. Normal ranges are 1.7 to 4.2 minutes in the dog, and 1.4 to 2.4 minutes in the cat. The bleeding time reflects in vivo primary hemostasis. It is prolonged with thrombocytopenia, thrombopathia, and vasculopathy. It is indicated in patients with a suspected primary hemostatic defect when the platelet count is adequate, and in the preoperative screening of patients considered at risk for von Willebrand disease or other thrombopathies. The buccal mucosal bleeding time is influenced by hematocrit and blood viscosity, and it has large interoperator and intraoperator variability (up to 2 minutes) in human beings and dogs.218,310 Prothrombin time (PT) and activated partial thromboplastin time (aPTT) assess secondary hemostasis via reagents that activate coagulation through the extrinsic or intrinsic pathway, respectively (see Figure 7-2).12 Prolongation of the PT indicates defective extrinsic and/or common pathways, whereas aPTT prolongation indicates defective intrinsic and/or common pathways. With isolated deficiency of a single factor, prolongation of the PT or aPTT generally does not occur until the factor is decreased to less than 25% to 30% of normal concentrations.12 Because of the short half-life of factor VII, the PT is very sensitive to vitamin K deficiency or antagonism. It is less sensitive to heparin than is the aPTT. A point-of-care coagulometer (SCA 2000, Synbiotics, San Diego, CA) is marketed for PT and aPTT testing in animals, with the use of nonanticoagulated or citrated whole blood. The latter provides superior sensitivity and specificity. Although point-of-care testing is invaluable, it is not equivalent to conventional laboratory testing, and it behooves the clinician to be aware of the limitations because they do influence interpretation. In canine patients, when the technique was compared with laboratory testing, sensitivities of the aPTT and PT were 100% and 86%, respectively; specificities were 83% and 96%, respectively.351 In the authors’ experience, the point-of-care coagulometer reliably detects significant defects; marked prolongations are usually clinically significant, and mild prolongations should be interpreted with caution. Results that do not correlate with clinical findings should be verified via conventional testing. Several commercial latex agglutination kits have been validated for use in dogs, and appear to have similar sensitivities and specificities.333 A single study comparing a serum-based assay (Thrombo-Wellcotest, International Murex Technologies Corporation, Toronto, Ontario, Canada) versus a plasma-based assay (FDP Plasma, American Bioproducts Incorporated, Parsippany, NJ) suggested that the latter may be more sensitive in the dog.39 Elevated fibrin split products concentrations are commonly detected with disseminated intravascular coagulation (DIC), but are not specific for the condition; elevated concentrations are also described in dogs with thromboembolism, neoplasia, immune-mediated hemolytic anemia, hepatic failure, sepsis and the systemic inflammatory response syndrome (SIRS), heatstroke, trauma, gastric-dilatation volvulus, and heart failure.39 D-Dimer is a neo-epitope produced when soluble fibrin is cross-linked by fXIIIa.24 The D-dimer epitope is exposed by plasmin-induced cleavage of cross-linked fibrin. In contrast to other fibrin split products, which indicate only the activation of plasmin, D-dimers indicate the activation of thrombin and plasmin, and are specific for active coagulation and fibrinolysis.334,335 Several D-dimer assays have been validated in dogs, including the semiquantitative Accuclot D-dimer latex agglutination assay (Sigma Chemical Company, St Louis, MO), the immunometric point-of-care NycoCard assay (Axis-Shield PoC, Abbott Park, IL), the Tina-Quant immunoturbidometric D-dimer assay (Boehringer Ingelheim Vetmedica Incorporated, St Joseph, MO), and the canine-specific immunochromatographic AGEN canine D-dimer test (Sigma Chemical Company).* The latex agglutination test is validated for feline use.344 Assay must be considered when interpreting results, because methods used may not be comparable. Moreover, few human assays will cross-react with canine or feline D-dimer, so it is important to ensure that the test has been validated in the species. D-Dimers are a sensitive indicator of thrombotic conditions, such as DIC and thromboembolism, and are more sensitive to thrombosis than are fibrin split products.39,262 They have excellent negative predictive value in that few dogs with DIC or thromboembolism have normal D-dimer concentrations.136,262 D-Dimers, however, are not specific; elevated concentrations are demonstrated in dogs with disseminated intravascular coagulation, thromboembolism, neoplasia, hepatic disease, renal failure, cardiac failure, or internal hemorrhage, and following surgical procedures.136,202,262,334 The diagnostic utility of D-dimers in cats remains uncertain. Specificity and sensitivity were low (56% and 67%, respectively) when cats with DIC were compared with cats suffering from various other conditions.344 Thromboelastography (TEG) enables global assessment of the hemostatic system in whole blood. The viscoelastic properties of the blood clot are evaluated, from initiation of coagulation, through amplification and propagation, to fibrinolysis.228 Information is generated regarding the strength and stability of the clot and the dynamics of its formation and breakdown. Compared with routine hemostatic tests, TEG provides a global assessment of hemostasis as determined by the interplay of plasma and cellular components.228 As such, it more closely reflects in vivo hemostasis. For facilities with TEG equipment, the test is not technically difficult, and results are available within 1 to 2 hours. TEG analysis is performed using a computerized thromboelastograph (Haemoscope Corporation, Niles, IL).101,371 The apparatus consists of a plastic cup and a pin suspended by a torsion wire. A sample of blood is placed in the cup (at 37° C) and the cup is elevated such that the pin hangs in the sample. The cup is then oscillated through an angle of 4°45” around the vertical axis. When fibrin strands form between the pin and the cup, the pin begins to move with the cup and the torque generated is transmitted to a transducer, which converts the signal data for computer display of the TEG tracing (Figure 7-7). Although TEG can be performed using fresh or citrated whole blood, the latter is invariably used in veterinary medicine; this requires recalcification of the sample immediately before testing.371 Testing is routinely performed at 30 minutes post sampling. Reliable and reproducible results can also be obtained at 120 minutes, but results are statistically different from 30 minutes.371 A strict time period from sampling to testing must be adhered to, and results compared with normal values established at that institution for that time period. Reliability is demonstrated in dogs with native citrated blood samples, and following activation with human recombinant tissue factor.* TEG can be performed using heparinase-impregnated cups, which inactivate heparin in the sample. This allows for accurate evaluation of catheter-drawn samples, as well as evaluation of the underlying coagulation status of heparinized patients. Many values can be derived from a TEG tracing (see Figure 7-7).349 The reaction time (R) represents the enzymatic portion of coagulation (secondary hemostasis). The clotting time (K) represents clot kinetics, largely determined by clotting factors, fibrinogen, and platelets. The angle (α) is dependent largely on fibrinogen, as well as on platelets and factors. The maximum amplitude (MA) represents the ultimate strength of the fibrin clot, dependent primarily on platelet aggregation (platelet number and function) and, to a lesser extent, on fibrinogen. The MA is used to derive the clot shear elastic modulus G, where G = 5000 × MA/(100 − MA), and is a measure of the overall coagulant status. TEG is used in human patients to identify hypocoagulability and hypercoagulability, to predict bleeding or thromboembolism, to guide transfusion therapy, and to monitor the impact of various therapeutic agents.123,175,184,321,349 TEG is rapidly gaining popularity in veterinary medicine. Utility has been reported to identify hypercoagulability and hypocoagulability in various canine disorders.† A prospective study evaluated the correlation between TEG and clinical signs of bleeding in dogs, compared with routine coagulogram.372 TEG correctly identified bleeding, with a positive predictive value of 89% and a negative predictive value of 98%, based on G alone. In contrast, the routine coagulogram had a positive predictive value of 50% to 81% and a negative predictive value of 92% to 93%. Additional studies are needed to determine the clinical utility of TEG in predicting bleeding or thromboembolic complications, and in guiding therapy. TEG has been reported in normal cats.7 Reliable results are commonly obtained, but assays can be complicated by unstable tracings and poor reproducibility. Tissue factor activation may help to overcome this effect. The utility of TEG in veterinary practice is limited by the few institutions that currently have the technology and by the need to run tests shortly after sampling. 1. Inadequate repair of vessels or vascular structures that are knowingly opened or divided, whether purposefully or accidentally 2. Occult or undiagnosed, and thus unrepaired, injury to the vascular system 3. Damage during the course of surgery to organs or structures within the operative field 4. Damage during the course of surgery, or in the immediate postoperative period, to organs or structures remote from the surgical site275
Bleeding and Hemostasis
ADP
Adenosine diphosphate
aPTT
Activated partial thromboplastin time
ASA
American Society of Anesthesiologists
AT
Antithrombin
COX
Cyclooxygenase
CTPA
Computed tomographic pulmonary angiography
DDAVP
De-amino D-arginine vasopressin, desmopressin
DIC
Disseminated intravascular coagulation
EACA
Epsilon-aminocaproic acid
FeLV
Feline leukemia virus
FIV
Feline immunodeficiency virus
IL
Interleukin
PAI
Plasminogen activator inhibitor
PGI2
Prostacyclin
PT
Prothrombin time
rFVIIa
Recombinant activated fVII
SIRS
Systemic inflammatory response syndrome
TCT
Thrombin clot time
TEG
Thromboelastography
TNF
Tumor necrosis factor
t-PA
Tissue-type plasminogen activator
u-PA
Urokinase-type plasminogen activator
vWF
von Willebrand factor
Hemostasis and Fibrinolysis
Primary Hemostasis
Secondary Hemostasis
A Cell-Based Model of Coagulation
Regulation of Hemostasis
Fibrinolysis
Hemostatic Testing
Platelet Enumeration and Estimation
Buccal Mucosal Bleeding Time
Prothrombin Time and Activated Partial Thromboplastin Time
Fibrin Split Products
D-Dimers
Thromboelastography
Bleeding
Causes of Surgical Bleeding
Technical Causes
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Bleeding and Hemostasis
Only gold members can continue reading. Log In or Register to continue
