Brain tumours occur in dogs more frequently than any other domestic species with a reported incidence of 14.5/100 000 in one US study (Vandevelde 1984). They are typically seen in older dogs (median age 9.5 years). An increased incidence of brain tumours has been reported in Boxers, Boston Terriers, Dobermans and Golden Retrievers (Heidner et al 1991). Brachycephalic breeds, e.g. Boxers, are more likely to develop glial cell tumours whilst dolichocephalic breeds are more likely to develop meningiomas (Summers et al 1995). The aetiology of canine brain tumours is unknown.
Pathogenesis
Brain tumours cause dysfunction either by primary destruction of brain tissue or by compression of adjacent anatomical structures. Increased intracranial pressure is often seen; this is due primarily to oedema and less commonly to haemorrhage or infarction.
As a rule, primary brain tumours grow slowly and therefore have a chronic clinical progression; in many cases this is seen by the client as normal slowing down due to age. Clinical signs are usually only seen when the brain can no longer compensate for the presence of the tumour, and in these cases the onset of neurological signs can be acute and severe. The most common clinical sign seen in patients with intracranial neoplasia is seizures. Therefore, in any older patient presenting with seizures, a primary differential should be a brain tumour. As these tumours are usually advanced at the time of diagnosis, immediate diagnostic evaluation is essential.
Primary brain tumours are usually solitary, but multiple tumours have been reported. The lack of a well-developed lymphatic system within the brain means that major patterns of spread involve local invasion and cerebrospinal fluid (CSF) seeding (LeCouteur & Withrow 2007). Multiple tumours of different histological types may occur rarely. Extracranial metastasis of primary brain meningiomas has been reported, but is considered rare (Schulman et al 1992).
Secondary tumours of the brain are seen due to metasta-tic spread, e.g. mammary carcinomas, haemangiosarcomas (HSA), prostatic and pulmonary carcinomas (LeCouteur & Withrow 2007). Of these malignancies HSA is seen most frequently and usually when there is evidence of metastasis to brain other extracranial sites are also involved (Snyder et al 2008). CNS lymphoma can occur solely within the CNS or as a secondary tumour late in the course of multicentric lymphoma or simultaneously with the initial presentation of multicentric lymphoma.
Other secondary tumours affect the brain by means of direct local extension, e.g. nasal, frontal sinus, skull, cranial nerve sheath tumours (e.g. oculomotor, vestibulocochlear and trigeminal nerves) or pituitary tumours (see Chapter 26). Skull tumours that affect the brain by local extension include osteosarcoma, chondrosarcoma and multilobular osteochondrosarcoma (LeCouteur & Withrow 2007).
Pathology
Gliomas and meningiomas are the most commonly recognized intracranial neoplasms of dogs, occurring with similar frequencies (Snyder et al 2006).
Glial cell neoplasms are classified based on the predominant cell type (e.g. astrocytoma, oligodendrocytoma). They are often found in the parenchyma of the brain, and can be very invasive.
Meningiomas arise from the arachnoid layer of the meninges; they are peripherally located and most commonly broad based and extra-axial (arise outside of and push into the brain parenchyma) (Bagley 2005a). Canine meningioma is benign, but locally infiltrates along the Virchow–Robin space and invariably lacks demarcation from normal brain tissue. In cats, meningiomas are almost always well defined and have a clear demarcation (LeCouteur & Withrow 2007). The growth rate of meningiomas in cats is slow compared to dogs.
Tumours of the ependyma and choroid plexus include choroid plexus papillomas, which occur relatively infrequently.
CNS lymphoma can be either primary or secondary. Histiocytic lymphoma or neoplastic reticulosis occurs as single or multiple mass lesions characterized histologically by a perivascular proliferation of ‘reticulohistiocytic cells’ with different patterns of reticulin production, admixed with varying populations of inflammatory cells (LeCouteur & Withrow 2007).
Embryonal tumours of the CNS/primitive neuroectodermal tumours are all derived from a germinal neuroepithelial cell that has the potential to differentiate along a number of neuroectodermal cell lines (neuronal, ependymal, glial). All have an anaplastic nature and are biologically malignant (LeCouteur & Withrow 2007).
Neuronal and mixed neuronal–glial neoplasms (gangliocytoma, ganglioglioma) occur in young animals.
Primary CNS lesions reported rarely include pineal tumours, germ cell tumours (germinoma, teratoma), primary CNS melanoma, chordoma, hamartoma, epidermoid and dermoid cysts and granular cell tumour (LeCouteur & Withrow 2007).
Clinical signs
The most common presenting clinical sign for a patient with a primary brain tumour is seizures (Bagley et al 1999, Snyder et al 2006). Changes in mentation, pacing, circling, head pressing, etc. are also common presenting signs. Other clinical signs can be seen depending on the location of the tumour within the CNS and associated secondary effects of the tumour (oedema, compression of normal brain tissue and increasing intracranial pressure). For example, neoplasms of the cerebellomedullary angle affect the vestibular system, causing vestibular signs, often with brain stem signs (paresis or loss of conscious proprioception), changes in mental status, and deficits in the fifth or seventh cranial nerve. Vertical nystagmus in any head position is most consistent with central vestibular disease. In addition, the nystagmus may change with changing head position (LeCouteur 2003).
Primary brain tumours are typically slow growing. Gradual changes in the individual may have been noticed by the client but attributed to advancing age. This means that the patient has been able to cope for a period of weeks to months before the onset of clinical signs that can be rapidly progressive due to the large size of the majority of tumours at the time of diagnosis. In any patient where a brain tumour is suspected, early diagnostic evaluation is encouraged as early diagnosis will undoubtedly contribute to longer survival times for patients and increase the treatment options available for them. It is essential, therefore, that for any middle-aged to older patient, advanced imaging is recommended. It is to be remembered that the prevalence of epilepsy as a cause of seizures declines rapidly after 5 years of age (Bagley 2005b).
Tumours that grow rapidly or obstruct a major blood vessel may show more acute signs due to haemorrhage or infarction. Metastatic lesions in the brain often lead to acute, rapidly progressive neurological signs. Localized clinical signs are seen most frequently and result from compression, invasion or irritation of a region of the brain, whereas generalized non-localizing signs result from secondary effects, e.g. increasing intracranial pressure (ICP).
Diagnostic work-up
A full neurological examination is required, but in many patients this may be unremarkable. In one study (Snyder et al 2006), 20% of primary intracranial tumours identified were in the olfactory area, an area of the brain that is often associated with a normal neurological examination in spite of the presence of a space-occupying lesion. An ophthalmic examination may be helpful in some patients with increased ICP because papilloedema (optic nerve oedema) may be present. If papilloedema is present, the risk of herniation is increased if CSF collection is planned. It should be remembered that papilloedema could also be seen with inflammatory disease of the CNS.
As with any older patient a minimum database should be established. Haematology, serum biochemistry and urinalysis may assist diagnosis of a primary extracranial malignancy (e.g. pancreatic insulinoma). Thoracic radiographs and abdominal ultrasound are recommended to ensure there is no evidence of extracranial neoplasia or other underlying problems (Snyder et al 2006). Rarely do primary intracranial tumours metastasize, but in patients with multifocal clinical signs, metastatic disease from another tumour should be ruled out.
Imaging, either MRI or CT, is required for definitive diagnosis (Figure 24.1). MRI is superior to CT in the evaluation of intracranial neoplasia. In the majority of cases intracranial neoplasms are not amenable to biopsy and the diagnosis is usually presumptive based on the imaging characteristics of the tumour and location within the CNS. CSF analysis is a secondary non-specific test but changes in CSF have been associated with intracranial tumours, the most common change being inflammatory. A neutrophilic pleocytosis has been associated with meningiomas (Bailey & Higgins 1986) but was not found to be a typical finding in another study (Dickinson et al 2006). The greatest elevations in CSF total protein were associated with choroid plexus tumours (Bailey & Higgins 1986). In patients with lymphoma, neoplastic cells may be identified. The value of CSF analysis is to rule out inflammatory and infectious causes of cerebral dysfunction rather than diagnose a brain tumour. Nevertheless, CSF analysis may support the diagnosis of a tumour; however, if a tumour is suspected, sampling should be taken after the MRI/CT scan as increased intracranial pressure may lead to brain herniation.
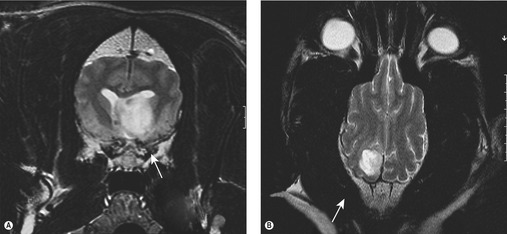
Differential diagnoses include vascular disorders (vascular accidents), trauma, immunological (e.g. granulomatous meningoencephalitis, GME), infection (e.g. protozoal), toxicity, degeneration, congenital or idiopathic (epilepsy).
MRI/CT characteristics of brain tumours
Meningiomas are usually broad-based, peripherally located and show uniform enhancement with contrast agent (Troxel et al 2004, Turrel et al 1986a).
Glial tumours typically are centrally located with ring-like, non-uniform enhancement and poorly defined margins (Kraft et al 1997, Troxel et al 2004, Turrel et al 1986a).
Choroid plexus tumours are well-defined, hyperdense masses with uniform contrast enhancement (Turrel et al 1986a).
Pituitary tumours are located at the sella turcica, have little peritumoral oedema and extend dorsally with uniform contrast enhancement and well-defined margins (Turrel et al 1986a).
Biopsy
The majority of brain tumours are ‘diagnosed’ based on the MRI/CT image, primarily because of the concerns about the risks of obtaining an ante-mortem diagnosis in a veterinary patient. If a histological diagnosis is obtained it is usually an excisional biopsy acquired if the tumour is considered resectable based on imaging studies. CT-guided stereotactic biopsy systems provide rapid and accurate means of obtaining a biopsy with a low complication rate; however, access to this technology is extremely limited and expensive.
Treatment options for canine brain tumours
Initial treatment
In all patients with primary brain tumours it is essential to control the secondary effects of the tumour, e.g. peritumoral oedema, seizures and increases in ICP pressure. Steroids are indicated to reduce oedema; in the seizuring patient anticonvulsant therapy should be started (phenobarbital or potassium bromide).
Medical management
For patients treated symptomatically only, survival times are in the range of 2–3 months. In one study the median survival time (MST) was only 6 days (Heidner et al 1991) for dogs with no treatment or symptomatic treatment. Obviously the overall survival time in an individual depends on a number of factors: type of tumour (patients with meningiomas may live longer as these tumours are slow growing), location within the brain, associated clinical signs and the ease with which they can be controlled, and the general health of the patient.
Surgery
Surgical options include complete removal, partial removal or biopsy. In dogs, meningiomas are more invasive than their feline counterparts but long-term survival with surgery, either as primary treatment or in combination with radiotherapy, has been reported. In one study, dogs treated with surgery alone had an MST of 198 days with a 1-year survival rate of 30% (Niebauer et al 1991). Survival times in excess of 2 years have been reported with combinations of surgery and radiotherapy (Heidner et al 1991).
In 31 dogs with intracranial meningiomas, those that underwent tumour resection alone and survived >1 week after surgery had an MST of 7 months (range 0.5–22 months). Dogs that underwent tumour resection followed by radiotherapy had an MST of 16.5 months (range 3–58 months) (Axlund et al 2002). Twenty dogs with incompletely resected intracranial meningiomas were treated with adjuvant radiotherapy and the 2-year progression-free survival was 68% (Théon et al 2000).
Removal of neoplasms in the caudal fossa and brain stem involves significant morbidity and mortality, and are better treated with other forms of therapy such as radiotherapy and/or chemotherapy. Calvarial tumours such as osteosarcoma, chondrosarcoma and multilobular osteochondrosarcoma (MLO) may be treated with surgical excision successfully (Dernell et al 1998) (see Chapter 21). Surgical debulking and adjuvant radiotherapy has been described as a treatment option for MLO (Dernell et al 1998, Straw et al 1989).
Greco et al (2006) reported on the use of a surgical aspirator to resect intracranial meningiomas in 17 dogs. Better outcomes were achieved than with traditional surgery alone or surgery and radiotherapy, or radiotherapy alone. The MST was 1254 days. Histological subtype of the tumour was prognostic (anaplastic, 0 days; fibroblastic, 10 days; psammomatous, >313 days; meningothelial, >523 days; transitional, 1254 days).
Radiotherapy
The treatment of choice for the majority of patients with a brain tumour is radiotherapy (Bley et al 2005, Evans et al 1993, LeCouteur et al 1987, Turrel et al 1986b). In one study (Bley et al 2005) 46 dogs with brain tumours were treated with a fine fractionated protocol (24 Gy/fraction to a total dose of 3552 Gy) had on MST of 1174 dogs. In contrast, Brearley et al (1999) repeated survival times for dogs treated with a hypo-fractionated protocol (streatments once weekly to a total dose of 41 Gy) of 49. 7 weeks for extra-avoid ones. As the majority of these tumours are large when first diagnosed, it is important that these patients are referred early to a facility equipped to treat them. The more compromised the patient, the poorer the prognosis, and as access to MRI and CT is becoming routine, making diagnosis easier and facilitating treatment as soon as possible, this will contribute to improve survival times as will the development of better treatment protocols. The authors advise prioritizing patients with brain tumours because of how quickly they can destabilize.
Side effects from radiotherapy treatment
Normal brain tissue is relatively resistant to the effects of radiotherapy, however, as with any treatment side effects can occur.
• Acute: These occur during or shortly after radiotherapy and are due to radiation-induced oedema. Initially, this may cause a mild increase in neurological signs; however, it is self-limiting and to minimize this patients undergoing radiotherapy for brain tumours can be given anti-inflammatory doses of prednisolone.
• Early-delayed: Early-delayed effects result from temporary demyelination caused by the effects of radiation on oligodendroglial cells or radiation-induced changes in vascular permeability. Early vascular abnormalities and tumour necrosis may induce clinical and radiographic signs that cannot be distinguished from tumour progression. Treatment is steroid therapy as required.
• Late-delayed: These effects, should they be seen, are potentially more serious; however, they are not seen until many months to years after treatment and will present as focal or diffuse damage to the white matter. The presentation depends on the extent of the damage and the exact location. This is attributed to vascular injury or to a direct effect on glial cells.
The actual tolerance of the brain depends on the total dose of radiation given and the dose/fraction. In the majority of patients treated with radiotherapy, as this is palliative therapy only, the recurrence of clinical signs due to tumour progression is more frequently seen than any late-delayed effects of treatment.
It should be noted that any patient whose initial clinical sign is seizures might require anticonvulsant therapy for the remainder of their life. Once a seizure focus has formed, then even with successful treatment of the underlying tumour, the patient may continue to have seizures if not on anticonvulsants. The authors recommend continuing anticonvulsant medication for at least 6 months after treatment. If at that time no seizures have occurred, it can be slowly withdrawn.
Chemotherapy
In humans, metastases to the CNS occur in ~25% of patients with systemic neoplasms. In veterinary patients the incidence appears to be low but this may be underestimated and the longer survival times that we now see in our veterinary patients may lead to more CNS metastases being recognized. The blood–brain barrier (BBB) limits entry of tumour cells into the brain, the exception being lymphoid tumours as lymphocytes can cross the BBB. However, it is thought that brain tumours may compromise the BBB and allow chemotherapy to access the tumour. Dogs with glial cell tumours and meningiomas may respond partially to lomustine and prednisolone (Jung et al 2006), or carmustine (Dimski & Cook 1990), or lomustine alone (Fulton & Steinberg 1990).
Metastatic disease to the CNS has a poor prognosis, and although radiotherapy can be considered as a palliative, this has not been widely reported.
CNS lymphoma overall carries a poor prognosis. Whole brain radiation will produce short-term control but in general CNS lymphoma is an aggressive tumour with a predilection for the leptomeninges.
Tumours of the spinal cord
Primary tumours of the spinal cord are seen infrequently in dogs and are classified as extradural (50%), intradural–extramedullary (30%) or intramedullary (15%) (Wright 1985). The median age is 10 years, with 30% of tumours occurring in patients less than 3 years of age (Luttgen et al 1980). The primary spinal cord neoplasm seen most frequently in young dogs is neuroepithelioma (Moissonnier & Abbott 1993). Ninety per cent of spinal cord tumours occur in large breed dogs (Luttgen et al 1980).
• Extradural tumours are usually primary malignant bone tumours (OSA, CSA, FSA, HSA, haemangioendothelioma, myeloma) and tumours metastatic to bone and soft tissue (Wright 1985)
• Intradural–extramedullary lesions are mostly meningiomas and peripheral nerve sheath tumours (Wright 1985). Meningiomas are the most commonly diagnosed primary spinal cord neoplasm in dogs. Fourteen per cent of canine CNS meningiomas involved the spinal cord, with the cervical cord most frequently affected (40% cervical, 32% thoracic, 28% lumbar) (Luttgen et al 1980). Peripheral nerve sheath tumours (PNSTs) also affect the spinal cord (Fingeroth et al 1987, Wright 1985). Neuroepithelioma (ependymoma, medulloepithelioma, nephroblastoma and spinal cord blastoma) is seen in young dogs (6 months to 3 years of age), especially German Shepherds and Retrievers. It has a predilection for T10–L2 spinal cord segments (Blass et al 1988, Ferretti et al 1993, Moissonnier & Abbott 1993, Ribas 1990, Summers & deLahunta 1986, Summers et al 1988, Tamke & Foley 1987).
• Intramedullary spinal tumours of dogs occur infrequently and are mostly of glial cell origin. Granulomatous meningoencephalitis (GME) may also affect the spinal cord. Metastatic lesions of HSA and lymphoma have a tendency for intramedullary spinal cord involvement (Waters & Hayden 1990). HSA is the most common secondary tumour affecting the canine spinal cord.
History and clinical signs
Extramedullary tumours are often slow growing, gradually resulting in spinal cord compression (weeks or months), but can show an acute onset of signs and are often painful. Intramedullary lesions tend to grow quickly, causing necrosis, ischaemia or haemorrhage, and corresponding rapid neurological dysfunction, without apparent pain.
Intradural–extramedullary tumours frequently have prolonged, intermittent signs. They are usually not painful until advanced and signs are often lateralized for PNST. Isolated plasmacytomas have also been identified.
Brachial or lumbar intumescence tumours may show lameness, root signature, muscle wasting and lower motor neuron (LMN) signs to the affected limb (see below).
Diagnostic workup
This comprises physical examination (including a neurological examination to localize the affected segment of the spinal cord), haematology, biochemistry, thoracic radiographs for primary or metastatic neoplasia, radiographs of vertebral column (best under general anaesthesia), CSF tap, myelography or advanced imaging (CT/MRI), and very occasionally a biopsy of the lesion.
Radiography
Primary or secondary vertebral tumours may produce bone lysis, new bone production or both. The vertebral body and arch are more often affected than dorsal spinal processes or transverse processes. Expansion of a spinal tumour may result in enlargement of an intervertebral foramen, widening of vertebral canal or thinning of surrounding bone.
CSF analysis
Lymphoma often results in an elevated white cell count that consists of predominantly abnormal lymphocytes (Lane et al 1994, Spodnick et al 1992).
Myelography
Myelography is a useful tool in the diagnosis of spinal cord neoplasms; however, it causes an inflammatory change in the CSF for up to 3 weeks so it is important to collect CSF before injection of contrast agent. Neurological function is often worse for 12–24 hours after myelography and the patient should be monitored closely for seizures that may occur after injection of contrast.
Advanced imaging
CT is useful for bone detail. CT and MRI are non-invasive and provide images preferable to radiography/myelography.
Biopsy
Surgical biopsy is usually not carried out due to morbidity, and often a presumptive diagnosis and therapeutic plan are made after a thorough work-up.
Treatment
The immediate goal is to relieve the deleterious effects of sustained spinal cord compression. Hemilaminectomy is the treatment of choice and, depending on the histological diagnosis, radiotherapy and/or chemotherapy may be indicated.
• Lymphoma: radiotherapy and chemotherapy.
• Isolated plasmacytomas: surgery and/or radiotherapy.
• Extradural sarcomas: palliative radiotherapy for bone pain; depending on neurological dysfunction, surgical decompression.
• Meningiomas: slow growing and can often be completely removed surgically with a good prognosis (Levy et al 1997). Adjuvant radiotherapy has been shown to increase survival times in patients with incompletely resected spinal cord tumours up to 20 months (Siegel et al 1996).
• Nerve sheath tumours: once the spinal cord is compromised, complete surgical removal may be difficult, and although adjuvant radiotherapy may help prolong the disease-free interval (Siegel et al 1996), the number of cases that have been managed is small.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


