Chapter 1 WATER METABOLISM AND DIABETES INSIPIDUS
Water consumption and urine production are controlled by complex interactions between plasma osmolality, fluid volume in the vascular compart-ment, the thirst center, the kidney, the pituitary gland, and the hypothalamus. Dysfunction in any of these areas results in the clinical signs of polyuria and polydipsia. Vasopressin (antidiuretic hormone [ADH]) plays a key role in the control of renal water resorption, urine production and concentration, and water balance. In the presence of vasopressin and dehydration, the average dog and cat can pro-duce urine concentrated to or above 2300 mOsm/kg. In the absence of vasopressin or vasopressin action on the kidneys, the urine may be as dilute as 20 mOsm/kg.
PHYSIOLOGY OF WATER METABOLISM
OVERVIEW.
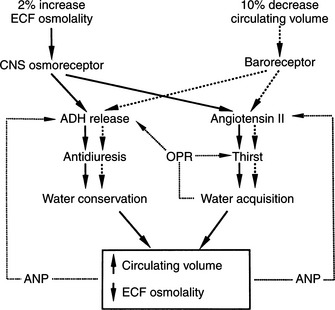
Plasma osmolality and its principal determinant, the plasma sodium concentration, are normally maintained within remarkably narrow ranges. This stability is achieved largely by adjusting total body water to keep it in balance with the serum sodium concentration. Water balance is controlled by an integrated system that involves precise regulation of water intake via thirst mechanisms and control of water output via stimulation of vasopressin secretion (Fig. 1-1). The major sources of fluid loss from the dog and cat include urine, the respiratory tract, and feces. As long as free access to water is allowed, total body water in hu-mans rarely varies by more than 1% to 2% (Aron et al, 2001). Some of the water necessary to maintain homeostasis is taken in with food; the majority is ingested as water.
The capacity of the kidney to produce concentrated urine plays an important part in maintenance of water balance. Animals eat a diet that produces osmotically active material ultimately excreted in urine, thus requiring water in which to be excreted. The more concentrated urine the kidney can produce, the less water is required to excrete those solutes.
THE NEUROHYPOPHYSIS.
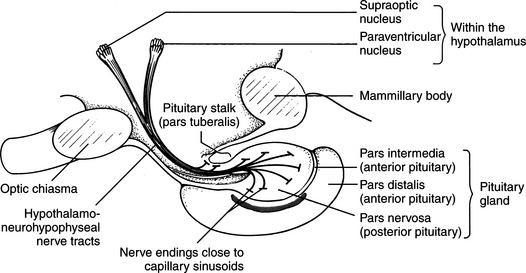
The neurohypophysis consists of a set of hypothalamic nuclei (supraoptic and paraventricular) responsible for the synthesis of oxytocin and vasopressin; the axonal processes of these neurons, which form the supraopticohypophysial tract; and the termini of these neurons within the posterior lobe of the pituitary (Fig. 1-2; Reeves et al, 1998). The neurosecretory cells in the paraventricular and supraoptic nuclei secrete vasopressin or oxytocin in response to appropriate stimuli. The neurosecretory cells receive neurogenic input from various sensor elements, including low-pressure baroreceptors located in the heart and arterial circulation and two circumventricular organs, the subfornical organ and the organum vasculosum of the lamina terminalis. These organs lie outside the blood brain barrier and may be important for osmoreception and interaction with blood-borne hormones, such as angiotensin II.
VASOPRESSIN: BIOSYNTHESIS, TRANSPORT, AND METABOLISM.
Vasopressin and oxytocin are nonapeptides composed of a six-membered disulfide ring and a three-membered tail on which the terminal carboxyl group is amidated (Fig. 1-3). Arginine vasopressin (AVP) is the antidiuretic hormone in all mammals except swine and other members of the suborder Suina, in which lysine vasopressin is synthesized (Reeves et al, 1998). Vasopressin differs from oxytocin in most mammals only in the substitution of phenylalanine for isoleucine in the ring and arginine for leucine in the tail. The ratio of antidiuretic to pressor effects of vasopressin is increased markedly by substituting d-arginine for l-arginine at position 8. This modification, as well as removal of the terminal amino group from cysteine, yields 1 deamino (8 d-arginine) vasopressin (DDAVP), a synthetic commercially available product (see Fig. 1-3). DDAVP is a clinically useful analogue with prolonged and enhanced antidiuretic activity that does not require injection to be effective.
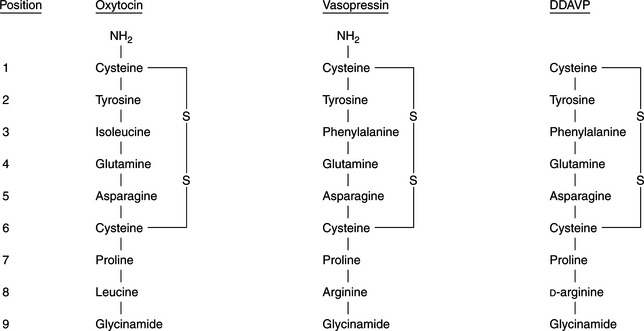
FIGURE 1-3 The chemical structures of oxytocin, vasopressin, and 1 deamino (8 D-arginine) vasopressin (DDAVP).
The production of vasopressin and oxytocin is associated with synthesis of specific binding proteins called neurophysins. One molecule of neurophysin I (estrogen-stimulated neurophysin) binds one molecule of oxytocin, and one molecule of neurophysin II (nicotine-stimulated neurophysin) binds one molecule of vasopressin (Reeves et al, 1998). The neurophysin peptide combination, often referred to as neurosecretory material, is transported along the axons of the hypothalamo-neurohypophyseal nerve tract and stored in granules in the nerve terminals located in the posterior pituitary gland (see Fig. 1-2). Release of vasopressin into the bloodstream occurs following electrical activation of the neurosecretory cells containing AVP. Secretion proceeds by a process of exocytosis, with release of vasopressin and neurophysin II into the bloodstream. In plasma, the neurophysin-vasopressin combination dissociates to release free vasopressin. Nearly all of the hormone in plasma exists in an unbound form, which because of its relatively low molecular weight, readily permeates peripheral and glomerular capillaries. Metabolic degradation of AVP appears to be mediated through binding of AVP to specific hormone receptors, with subsequent proteolytic cleavage of the peptide (Reeves et al, 1998). Renal excretion is the second method for elimination of circulating hormone and accounts for about one-fourth of total metabolic clearance.
ACTIONS OF VASOPRESSIN.
Cellular Actions.
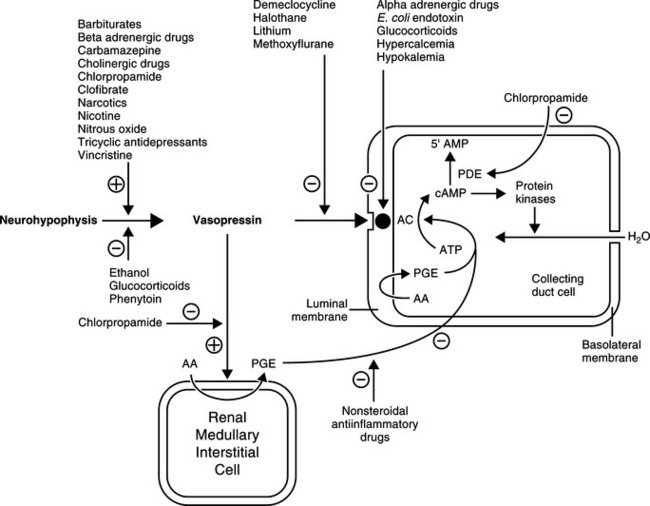
AVP acts via tissue receptors classified as V1 receptors in smooth muscle and V2 receptors in renal epithelia (Reeves et al, 1998). Only the latter receptors activate adenylate cyclase. The antidiuretic action of AVP is mediated through V2 cyclic AMP-dependent receptors, whereas its vasoconstrictive action is mediated through V1 phosphatidylinositol dependent receptors. Vasopressin stimulates V1 and V2 receptors, whereas the vasopressin analogue, desmopressin (DDAVP), which is commonly used for the treatment of central diabetes insipidus, has a strong affinity for V2 receptors with minimal pressor (V1) activity.
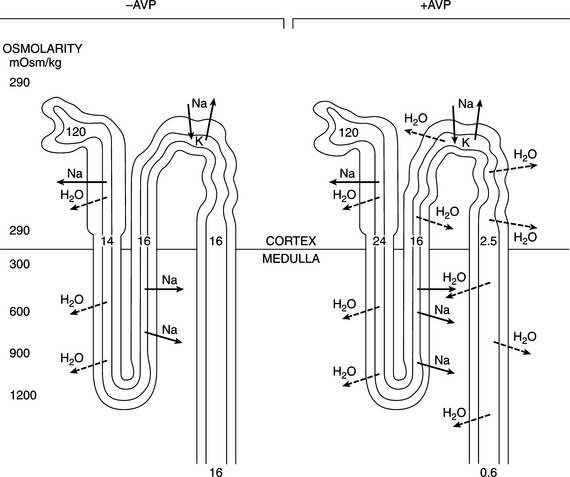
The major antidiuretic contribution of AVP is to increase the water permeability of terminal nephron segments or collecting ducts. The effects of AVP are mediated primarily by the intracellular second messenger cAMP (Fig. 1-4). AVP binds to the V2 receptors of hormone-responsive epithelial cells and activates membrane-associated adenylate cyclase to catalyze cAMP generation from ATP. cAMP-dependent activation of the protein kinase system leads to an increase in the water permeability of the luminal membrane of the cell as a result of insertion of aquaporin-2 water channels into the apical membrane of the epithelial cell. Transmembrane water movement occurs through these water channels, rather than by diffusion across the lipid bilayer or through junctional complexes (Fig. 1-5; Kanno et al, 1995; Lee et al, 1997). In essence, AVP, working via cAMP and protein kinase, alters water transport in hormone-responsive epithelia by causing the microtubule-dependent insertion of specialized membrane units (aquaporin-2 water channels) into the apical plasma membranes of these cells. The increase in water permeability in these segments augments osmotic water flow from the tubular lumen into a hypertonic medullary interstitium, thus providing for maximal urine concentration during antidiuresis (Reeves et al, 1998).
The intracellular concentration of cAMP appears to be the primary factor regulating the cellular actions of AVP. Increased concentrations of cAMP result from either enhanced formation (i.e., stimulation of adenyl cyclase following the interaction of AVP with receptors) or decreased catabolism. cAMP phosphodiesterase catalyzes the breakdown of cAMP to 5’AMP. Several drugs, hormones, and disease conditions change the renal tubular response to AVP by altering the interaction of AVP with its receptor, the activation of adenyl cyclase, or the catabolism of cAMP.
Clinical Effect.
The primary effect of AVP is to conserve body fluid by reducing the volume of urine production (Table 1-1). This antidiuretic action is achieved by promoting the reabsorption of solute free water in the distal and/or collecting tubules of the kidney. In the absence of AVP, the membranes lining this portion of the nephron are uniquely resistant to the diffusion of both water and solutes. Hence the hypotonic filtrate formed in the more proximal portion of the nephron passes unmodified through the distal tubule and collecting duct. In this condition, referred to as water diuresis, urine osmolality is low and urine volume is great (see Fig. 1-5).
TABLE 1-1 ACTIONS OF VASOPRESSIN
| Target Organ | Action |
|---|---|
In the presence of AVP and normal renal receptor activity, the hydro-osmotic permeability of the distal and collecting tubules increases, allowing water to back-diffuse down the osmotic gradient that normally exists between tubular fluid and the isotonic or hypertonic milieu of the renal cortex and medulla. Because water is reabsorbed without solute, the urine that remains within the lumen of the nephron has an increased osmotic concentration, as well as a decreased rate of flow through the tubules. The amount of water reabsorbed in the distal nephron depends on the plasma AVP concentration and the existence of a significant osmotic gradient in the renal interstitium. Vasopressin does not cause an active (i.e., energy-requiring) reabsorption of solute free water. It merely “opens the water channels” in the luminal membrane to allow water to flow in the direction of the higher osmolality (along the osmotic gradient). In the normal animal, the osmolality of the filtrate entering the distal tubule is low, whereas that of the renal interstitium is high, promoting reabsorption of water when the pores are open. Increasing the renal medullary interstitial osmolality increases the ability to reabsorb water and concentrate urine; thus desert rodents with extremely concentrated medullary interstitium can produce urine more concentrated than that of dogs and are remarkably capable of conserving fluid. Conversely, loss of the renal medullary hypertonicity may inhibit vasopressin’s antidiuretic activity (see Fig. 1-5). Decreased medullary hypertonicity (or lack thereof) can result from various causes, such as chronic water diuresis or reduced medullary blood flow. However, because a majority of fluid flowing from the loop of Henle can still be reabsorbed isotonically in the distal convoluted tubule and proximal collecting duct, loss of the hypertonic medullary concentration gradient alone rarely results in marked polyuria (Robertson, 1981).
It should be noted that 85% to 90% of the fluid filtered by the glomerulus is reabsorbed isosmotically with sodium and glucose in the proximal portion of the nephron. Sodium is then selectively reabsorbed from the remaining fluid, making the fluid hypotonic as it reaches the distal nephron. An additional 90% of this remaining fluid can be reabsorbed under the influence of AVP (Robertson, 1981). However, if the oral intake of salt is high or if a poorly reabsorbed solute such as mannitol, urea, or glucose is present in the glomerular filtrate, fluid resorption from the proximal tubule is impaired. The resultant increase in fluid volume presented to the distal nephron may overwhelm its limited capacity to reabsorb water. As a consequence, urine osmolality decreases and volume increases, even in the presence of large amounts of vasopressin. This type of polyuria is referred to as solute diuresis to distinguish it from that due to a deficiency of vasopressin action (see Complications of the Modified Water Deprivation Test, page 29). Conversely, in clinical situations such as congestive heart failure, in which the proximal nephron reabsorbs increased amounts of filtrate, the capacity to excrete solute free water is greatly reduced, even in the absence of vasopressin.
The physiologic significance of other vasopressin actions, listed in Table 1-1, is less clear. It has been suggested that the pressor actions of vasopressin are somehow important in the maintenance of blood pressure during hypovolemia. Vasopressin also acts on the gastrointestinal tract and the central nervous system (CNS). The neurophysins have no recognized biologic action apart from complexing oxytocin and vasopressin in neurosecretory granules of the neurohypophysis.
THIRST CENTER.
Consumption of water to preserve body fluid tonicity is governed by the sense of thirst, which in turn is regulated by many of the same factors that determine AVP release. The sensation of thirst is controlled by osmoreceptors located close to the AVP synthesizing cells in the hypothalamus. The specificities of the osmoregulation of thirst and of AVP release are similar (e.g., hypertonic NaCl stimulates both thirst and AVP release), whereas hypertonic urea or glucose stimulates neither (Reeves et al, 1998). In spite of the functional similarities of the osmoregulation of thirst and AVP secretion, electrophysiologic studies indicate that they are mediated by two distinct but adjacent osmoreceptors.
REGULATION OF THIRST AND VASOPRESSIN SECRETION.
Plasma Osmolality.
The most important stimulus for thirst and vasopressin secretion under physiologic conditions is plasma osmolality. At plasma osmolalities below a certain minimum or threshold value (approximately 280 mOsm/kg), plasma vasopressin is uniformly suppressed to low or undetectable levels. Above this point, plasma vasopressin and the sensation of thirst increase in direct proportion to increases in plasma osmolality (Fig. 1-6). The relationship among thirst, plasma AVP concentration, and plasma osmolality is quite sophisticated. Increases of as little as 1% in plasma osmolality result in stimulation of water intake and vasopressin secretion (Hammer et al, 1980).
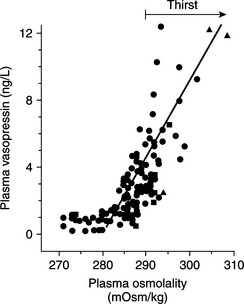
FIGURE 1-6 The relationship between plasma osmolality and plasma vasopressin level.
(Adapted from Robertson GL, Berl T: Water metabolism. In Brenner BM, Rector FC Jr (eds): The Kidney, 3rd ed. Philadelphia, WB Saunders, 1986, p 385.)
Studies indicate that an important role is also played by the blood-brain barrier, again suggesting unique permeability characteristics. Osmoreceptors appear to be situated in an area of the brain where the blood-brain barrier is deficient and are thus influenced by the composition of plasma rather than cerebrospinal fluid (Aron et al, 2001). Two circumventricular organs, the subfornical organ and the organum vasculosum of the lamina terminalis, lie outside the blood-brain barrier and are believed to be important for osmoreception, interaction with blood-borne hormones (e.g., angiotensin II), and regulation of AVP secretion by neurosecretory cells.
Blood Volume and Pressure.
Thirst and AVP secretion may be stimulated by contraction of the extracellular fluid volume without a change in plasma osmolality. Such an extracellular fluid loss may occur secondary to hemorrhage, for example, and results in both increased fluid consumption and vasopressin secretion. Small decreases in volume have little effect on AVP secretion, but any reduction exceeding 10% of the extracellular fluid causes marked stimulation that not only conserves water but may also be important in maintaining blood pressure (Aron et al, 2001).
Volume-mediated release of AVP may occur as a consequence of stimuli arising from “volume receptors,” or baroreceptors. Low pressure baroreceptors are located in the venous bed of the systemic circulation, the right side of the heart, and the left atrium, whereas high pressure baroreceptors are located within the systemic arterial system of the carotid sinus and aortic arch (Reeves et al, 1998). The electrical activity of the baroreceptor is related to the degree of stretch in the vessel wall. Increases in pressure and wall tension increase receptor firing rate, whereas decreases in blood pressure or blood volume decrease electrical activity. An inverse relationship exists between baroreceptor electrical activity and AVP secretion; that is, decreased electrical activity of the baroreceptor stimulates AVP secretion. The afferent pathways for the atrial and carotid bifurcation baroreceptors appear to be the vagus and glossopharyngeal nerves, respectively. Electrical activation of the thirst center and neurosecretory cells containing AVP is controlled by groups of neurons located in the anterior hypothalamus near, but distinct from, the supraoptic and paraventricular nuclei.
Interaction of Plasma Osmolality and Blood Volume.
Thirst mechanisms also involve interactions between extracellular fluid volume and osmolality. During periods of dehydration, increased plasma osmolality provides approximately 70% of the increased thirst drive, and the remaining 30% is due to hypovolemia. In salt depletion, the situation is less clear, but the normal drinking behavior or increased drinking observed in experimental animals has been attributed to the associated hypovolemia (Ramsay, 1983).
Miscellaneous Factors.
A variety of nonosmotic and nonhemodynamic factors may also stimulate AVP secretion. With varying potency, these factors include nausea, hypoglycemia, the renin angiotensin system, and nonspecific stress caused by factors such as pain, emotion, and physical exercise. A large number of drugs and hormones have also been implicated in the alteration of vasopressin secretion. This list includes agents that either stimulate or inhibit AVP secretion, as well as substances that potentiate or inhibit the renal tubular response to AVP (Table 1-2; see Fig 1-4; Reeves et al, 1998).
TABLE 1-2 DRUGS AND HORMONES REPORTED TO AFFECT VASOPRESSIN SECRETION OR ACTION
| SECRETION | |
|---|---|
| Stimulate AVP release | Inhibit AVP release |
| Acetylcholine | α-Adrenergic drugs |
| Anesthetic agents | Atrial natriuretic peptide |
| Angiotensin II | Glucocorticoids |
| Apomorphine | Haloperidol |
| β-Adrenergic drugs | Oxilorphan |
| Barbiturates | Phenytoin |
| Carbamazepine | Promethazine |
| Clofibrate | |
| Cyclophosphamide | |
| Histamine | |
| Insulin | |
| Metoclopramide | |
| Morphine and narcotic analogues | |
| Prostaglandin E2 | |
| Vincristine | |
| RENAL | |
|---|---|
| Potentiate AVP action | Inhibit AVP action |
| Aspirin | α-Adrenergic drugs |
| Carbamazepine | Atrial natriuretic peptide |
| Chlorpropamide | Barbiturates |
| Nonsteroidal anti-inflammatory agents | Demeclocycline |
| Glucocorticoids | |
| Thiazides | Hypercalcemia |
| Hypokalemia | |
| Methoxyflurane | |
| Prostaglandin E2 | |
| Protein kinase C | |
| Tetracyclines | |
| Vinca alkaloids | |
SATIATION OF THIRST.
Dehydrated animals have a remarkable capacity to consume the appropriate volume of water to repair a deficit. It has been demonstrated that dogs deprived of water for various periods of time drink just the volume of water needed to meet the deficit within 5 minutes. All animals have this capacity, although some species take longer to ingest the required amount of fluid. Satiation of thirst in dogs and cats requires restoration of normal plasma osmolality and blood volume, with correction of plasma osmolality playing the major role. In dogs with hypertonic volume depletion, restoration of osmolality in the carotid circulation without correcting osmolality outside the CNS caused a 70% decrease in drinking (Reeves et al, 1998). Restoration of blood volume in these dogs without ameliorating plasma hypertonicity reduced drinking by about 30%. Additional mechanisms may also play a minor role, including gastric distention and perhaps the participation of receptors in the liver. Similar inhibitory influences affect vasopressin secretion. Following voluntary rehydration in dehydrated animals, plasma vasopressin secretion returns to normal before redilution of the body fluids has been completed.
DIFFERENTIAL DIAGNOSES FOR POLYDIPSIA AND POLYURIA
Increased thirst (polydipsia) and urine production (polyuria) are common owner concerns in small animal veterinary practice. In dogs and cats, normal water intake varies from 20 to 70 ml/kg per day, and normal urine output varies between 20 and 45 ml/kg per day (Barsanti et al, 2000). Polydipsia and polyuria in the dog and cat have been defined as water consumption greater than 100 ml/kg/day and urine production greater than 50 ml/kg/day, respectively. It is possible, however, for individual dogs and cats to have abnormal thirst and urine production within the limits of these normal values. Polyuria and polydipsia usually exist concurrently, and determining the primary component of the syndrome is one of the initial diagnostic considerations when approaching the problem of polydipsia and polyuria (see page 13).
A variety of metabolic disturbances can cause polydipsia and polyuria (Table 1-3). These disorders can be classified, on the basis of underlying pathophysiology, into primary pituitary and nephrogenic diabetes insipidus; secondary nephrogenic diabetes insipidus resulting from interference with the normal interaction of AVP with renal tubular V2 receptors, generation of intracellular cAMP, or renal tubular cell function, or from loss of the renal medullary interstitial concentration gradient; osmotic diuresis-induced polyuria and polydipsia; or interference with hypothalamic/pituitary secretion of AVP.
TABLE 1-3 DIFFERENTIAL DIAGNOSIS FOR POLYDIPSIA AND POLYURIA AND USEFUL DIAGNOSTIC TESTS
| Disorder | Diagnostic Aids |
|---|---|
| Diabetes mellitus | Fasting blood glucose, urinalysis |
| Renal glycosuria | Fasting blood glucose, urinalysis |
| Chronic renal failure | BUN, creatinine, Ca:P, urinalysis |
| Postobstructive diuresis | History, monitoring urine output |
| Pyometra | History, CBC, abdominal radiography, abdominal ultrasonography |
| Escherichia coli & septicemia | Blood cultures |
| Hypercalcemia | Serum calcium |
| Hepatic insufficiency | Biochemistry panel, bile acids, ammonia tolerance test, abdominal radiography and ultrasonography |
| Hyperadrenocorticism | ACTH stimulation test, dexamethasone screening test, urine cortisol/creatinine ratio |
| Primary hyperaldosteronism | Serum sodium and potassium, blood pressure, abdominal ultrasonography, ACTH stimulation test (aldosterone) |
| Bacterial pyelonephritis | Urine culture, abdominal ultrasonography, excretory urography |
| Hypokalemia | Serum potassium |
| Hyponatremia | Serum sodium |
| Hypoadrenocorticism | Na:K, ACTH stimulation test |
| Hyperthyroidism | Serum thyroxine |
| Diabetes insipidus | Modified water deprivation test |
| Psychogenic polydipsia | Modified water deprivation test |
| Polycythemia | CBC |
| Acromegaly | Serum GH and IGF-I, CT scan |
| Paraneoplastic disorders | |
| Intestinal leiomyosarcoma | Abdominal ultrasonography, biopsy |
| Iatrogenic disorders | History |
| Very low protein diet | History |
Osmotic diuresis
PRIMARY RENAL GLYCOSURIA.
This uncommon disorder is seen primarily in the Basenji and Norwegian Elkhound. Primary renal glycosuria is a congenital renal tubular disorder resulting in an inability to reabsorb glucose from the ultrafiltrate in the nephron. In some dogs and cats, renal glycosuria may also be a component of a Fanconi-like syndrome, in which phosphate, potassium, uric acid, amino acids, sodium, and/or bicarbonate may also be inadequately reabsorbed from the ultrafiltrate. As in diabetes mellitus, glucose appears in the urine and acts as an osmotic diuretic, causing polyuria and, in turn, polydipsia. Urinalysis and fasting blood glucose measurement are sufficient initial screening tests for this disorder.
Vasopressin (antidiuretic hormone) deficiency
Partial or complete lack of vasopressin production by the neurosecretory cells located in the supraoptic and paraventricular nuclei in the hypothalamus is called central diabetes insipidus (CDI). This syndrome is discussed in subsequent sections (page 15).
Primary nephrogenic diabetes insipidus
A partial or complete lack of response of the renal tubule to the actions of AVP is called nephrogenic diabetes insipidus (NDI). Primary NDI results from a congenital defect involving the cellular mechanisms responsible for “opening the water channels” that allow water to be absorbed from the renal tubular ultrafiltrate. This syndrome is discussed in subsequent sections (page 17).
Acquired (secondary) nephrogenic diabetes insipidus
PYOMETRA.
Bacterial endotoxins, especially those associated with Escherichia coli, can compete with AVP for its binding sites on the renal tubular membrane, causing a potentially reversible renal tubular insensitivity to AVP. The kidneys have an impaired ability to concentrate urine and conserve water, and polyuria with compensatory polydipsia develops. Pyometra is the most common infectious disorder associated with the development of polyuria and polydipsia, although it has also been reported with prostatic abscessation, pyelonephritis, and septicemia (Barsanti et al, 2000). Affected bitches and queens may produce extremely dilute urine, causing fluid depletion and compensatory polydipsia. Normal urine-concentrating ability usually returns within days of successfully eliminating pyometra. The diagnosis of secondary NDI is presumptive in any polyuric/polydipsic bitch or queen with pyometra.
HYPERCALCEMIA.
Increases in serum calcium concentration may inhibit binding of AVP to its receptor site, damage AVP receptors in the renal tubules, inactivate adenyl cyclase and interfere with the action of AVP at the renal tubular level (acquired NDI), or decrease transport of sodium and chloride into the renal medullary interstitium. Polydipsia and polyuria are common early signs of hypercalcemia, which is easily diagnosed with a serum biochemistry panel. Once hypercalcemia is identified, the clinician must undertake an often extensive diagnostic evaluation to determine its cause (see Chapter 16).
HEPATIC INSUFFICIENCY AND PORTOSYSTEMIC SHUNTS.
Liver insufficiency and portosystemic shunts are recognized causes of polyuria and polydipsia. Many of the metabolic causes of polyuria and polydipsia (e.g., diabetes mellitus, hyperadrenocorticism, hypercalcemia) secondarily affect the liver, making it difficult to determine the role of the liver in causing polyuria and polydipsia. The exact cause of the polyuria is not known but may involve loss of medullary hypertonicity secondary to impaired urea nitrogen production or altered renal blood flow, increased GFR and renal volume, hypokalemia, impaired metabolism of cortisol, and primary polydipsia (Deppe et al, 1999). Urea nitrogen is a major constituent in the establishment and maintenance of the renal medullary concentration gradient. Without urea nitrogen the kidney loses the ability to concentrate urine, causing polyuria and compensatory polydipsia. Hepatic insufficiency and portosystemic shunts are usually suspected after evaluation of a complete blood count (CBC), serum biochemistry panel, urinalysis, and abdominal ultrasonography; these causes are confirmed with a liver function test (e.g., pre- and postprandial bile acids, ammonia tolerance test), specialized diagnostic imaging (e.g., positive contrast portogram, technetium scan) and histologic evaluation of an hepatic biopsy.
HYPERADRENOCORTICISM (CUSHING’S SYNDROME).
Polyuria and polydipsia are common clinical signs of hyperadrenocorticism. Glucocorticoids inhibit AVP release by a direct effect within the hypothalamus and/or neurohypophysis (Papanek and Raff, 1994; Papanek et al, 1997). This inhibition of AVP release is characterized by both an increase in osmotic threshold and a decrease in the sensitivity of the AVP response to increasing osmolality (Biewenga et al, 1991). Hyperadrenocorticism also causes resistance to the effect of AVP in the kidney, possibly through interference with the action of AVP at the level of the renal collecting tubules or direct depression of renal tubular permeability to water. In a few patients, a deficiency in AVP may result from direct compression of neurosecretory cells by an enlarging pituitary tumor. Suspicion of hyperadrenocorticism is usually aroused after careful review of the history, physical examination, and results of CBC, serum biochemistry panel, and urinalysis. Confirmation requires appropriate pituitary adrenocortical function tests (see Chapter 6).
PRIMARY HYPERALDOSTERONISM.
Polyuria and polydipsia have been reported in cats and dogs with primary hyperaldosteronism. The mechanism for polyuria and polydipsia is not clear, although mineralocorticoid-induced renal resistance to the actions of AVP and disturbed osmoregulation of AVP release has been documented in a dog with primary hyperaldosteronism (Rijnberk et al, 2001). Similar abnormalities have been identified in dogs with glucocorticoid excess, suggesting similar mechanisms of action for the polyuria and polydipsia in hyperaldosteronism and hyperadrenocorticism. The typical findings with primary hyperaldosteronism include weakness, severe hypokalemia, hypernatremia, systemic hypertension and adrenomegaly on abdominal ultrasound. Plasma aldosterone concentrations before and after ACTH administration are increased, and plasma renin activity is suppressed (see Chapter 6).
HYPOKALEMIA.
Hypokalemia is believed to render the terminal portion of the nephron less responsive to AVP, possibly by suppressing the generation of intracellular cAMP in renal tubular cells. Hypokalemia may also affect the hypertonic medullary interstitial gradient by interfering with solute accumulation and may interfere with release of AVP from the pituitary. Polyuria and polydipsia are not common clinical signs of hypokalemia. The most common clinical signs are related to neuromuscular dysfunction of skeletal, cardiac, and smooth muscle (e.g., weakness, cervical ventriflexion). Hypokalemia usually develops secondary to another disorder (Table 1-4), many of which also cause polyuria and polydipsia.
TABLE 1-4 CAUSES OF HYPOKALEMIA IN THE DOG AND CAT
Modified from DiBartola SP and De Morais HA: Disorders of potassium: Hypokalemia and hyperkalemia. In, DiBartola SP, editor: Fluid Therapy in Small Animal Practice, ed 2, Philadelphia, 2000, WB Saunders, p. 93. (source)
HYPOADRENOCORTICISM (ADDISON’S DISEASE).
Adrenocortical insufficiency results in impaired ability to concentrate urine (see Chapter 8). Despite normal kidney function and severe hypovolemia, most dogs with hypoadrenocorticism have a urine specific gravity of less than 1.030. Mineralocorticoid deficiency results in chronic sodium wasting, renal medullary solute washout, and loss of the medullary hypertonic gradient. Adrenalectomy in rats also decreases AVP-stimulated activation of renal medullary adenylate cyclase, primarily because of impairment in the coupling between the AVP receptor complex and adenylate cyclase. Treatment with dexamethasone corrects the defect. Hypercalcemia occurs in some patients with hypoadrenocorticism and may also play a role in the generation of polyuria and polydipsia.
ACROMEGALY.
Excessive secretion of growth hormone (GH) in the adult dog or cat results in acromegaly (see Chapter 2). Acromegaly causes carbohydrate intolerance and the eventual development of overt diabetes mellitus. In most cats and dogs with acromegaly, the polyuria is assumed to be caused by an osmotic diuresis induced by glycosuria. Renal insufficiency from a diabetic or GH-induced glomerulonephropathy may also play a role (Peterson et al, 1990).
POLYCYTHEMIA.
Polyuria and polydipsia may occur with polycythemia. Studies in 2 dogs with secondary polycythemia identified an increased osmotic threshold for AVP release, resulting in a delayed AVP response to increasing plasma osmolality (van Vonderen et al, 1997a). The authors attributed the abnormal AVP response to increased blood volume and hyperviscosity, which stimulate atrial natriuretic peptide (ANP) secretion and atrial and carotid bifurcation baroreceptors. ANP inhibits AVP release from the pituitary gland and the renal collecting duct’s responsiveness to AVP (Dillingham and Anderson, 1986; Lee et al, 1987).
Primary and psychogenic polydipsia
Primary polydipsia is defined as a marked increase in water intake that cannot be explained as a compensatory mechanism for excessive fluid loss. In humans, primary polydipsia results from a defect in the thirst center or may be associated with mental illness (Reeves et al, 1998). Primary dysfunction of the thirst center resulting in compulsive water consumption has not been reported in the dog or cat, although an abnormal vasopressin response to hypertonic saline infusion has been reported in dogs with suspected primary polydipsia (van Vonderen et al, 1999). A psychogenic or behavioral basis for compulsive water consumption does occur in the dog but has not been reported in the cat. Psychogenic polydipsia may be induced by concurrent disease (e.g., hepatic insufficiency, hyperthyroidism) or may represent a learned behavior following a change in the pet’s environment. Polyuria is compensatory to prevent overhydration. Psychogenic polydipsia is diagnosed by exclusion of other causes of polyuria and polydipsia and by demonstrating that the dog or cat can concentrate urine to a specific gravity in excess of 1.030 after water deprivation. This syndrome is discussed in more detail in subsequent sections (page 17).
Iatrogenic (drug-Induced) causes of polydipsia and polyuria
Several drugs have the potential to cause polyuria and polydipsia (Table 1-5). The most commonly encountered in small animal veterinary practice are glucocorticoids, diuretics, anticonvulsants (e.g., phenobarbital), synthetic levothyroxine, and salt supplementation. Drug-induced polyuria and polydipsia do not usually pose a diagnostic challenge. The polyuria and polydipsia should resolve following discontinuation of the drug. If polyuria and polydipsia persist, a concurrent disorder causing polyuria and polydipsia or renal medullary solute washout should be considered.
TABLE 1-5 DRUGS AND HORMONES CAUSING POLYURIA AND POLYDIPSIA IN DOGS AND CATS
Renal medullary solute washout
Loss of renal medullary solutes, most notably sodium and urea, results in loss of medullary hypertonicity and impaired ability of the nephron to concentrate the ultrafiltrate. Renal medullary solute washout is usually caused by one of the disorders previously described. It has also been associated with chronic diuretic therapy and abnormalities in circulation, such as hyperviscosity syndromes (polycythemia, hyperproteinemia), renal lymphatic obstruction (lymphosarcoma, lymphangiectasia), and systemic vasculitis (septicemia, systemic lupus erythematosus). Perhaps the most important clinical ramification of renal medullary solute washout is its potential to interfere with results of the modified water deprivation test (see page 32). Hypertonicity of the renal medulla is usually restored once the underlying cause of the polyuria and polydipsia is corrected.
DIAGNOSTIC APPROACH TO POLYURIA AND POLYDIPSIA
Assessment of urine specific gravity may be helpful in identifying polyuria and polydipsia and may provide clues to the underlying diagnosis, especially if multiple urine specific gravities are evaluated (Table 1-6). Urine specific gravity varies widely among healthy dogs and, in some dogs, can range from 1.006 to greater than 1.040 within a 24 hour period (van Vonderen et al, 1997b). Wide fluctuations in urine specific gravity have not been reported in healthy cats.
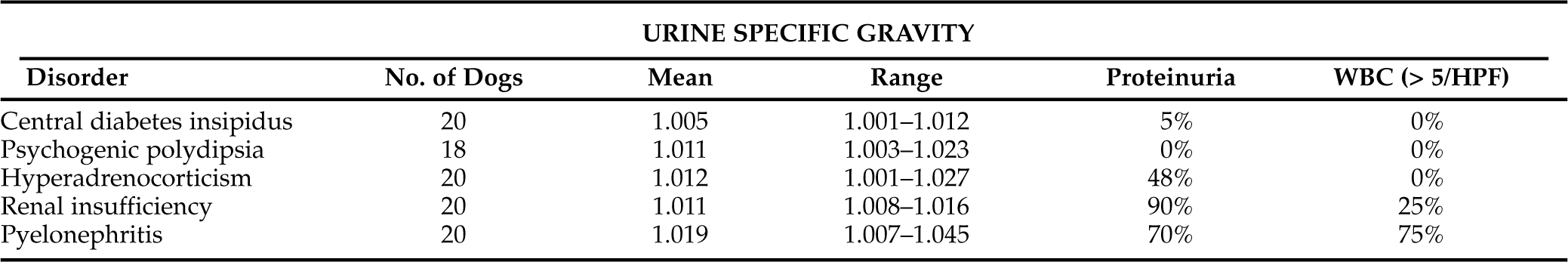
We prefer to have the owner collect several urine samples at different times of the day for 2 to 3 days, storing the urine samples in the refrigerator until they can be brought to the veterinary hospital for determination of urine specific gravity. Urine specific gravities measured from multiple urine samples that are consistently less than 1.030 (especially less than 1.020) support the presence of polyuria and polydipsia and the need for a diagnostic evaluation to determine the cause. Identification of one or more urine specific gravities greater than 1.030 supports normal urine concentrating ability and an intact, functioning pituitary vasopressin-renal tubular cell axis. Dogs and cats may still have polyuria and polydipsia despite identification of concentrated urine; possible differentials include disorders causing an osmotic diuresis (e.g., diabetes mellitus), psychogenic polydipsia and disorders in the regulation of AVP secretion (van Vonderen et al, 1999).
Many potential causes exist for the development of polyuria and polydipsia in dogs and cats (see Table 1-3), one of the least common being diabetes insipidus. An animal with a history of severe polydipsia and polyuria should be thoroughly evaluated for other causes of polydipsia and polyuria prior to performing specific diagnostic procedures for diabetes insipidus (Fig. 1-7). The array of differential diagnoses precludes premature or unsubstantiated formation of a diagnosis and treatment plan. It is necessary to establish a firm data base. Initial information allows inclusion or exclusion of the many common medical disorders associated with polyuria and polydipsia that are contrasted with the less common CDI, NDI, or psychogenic polydipsia.
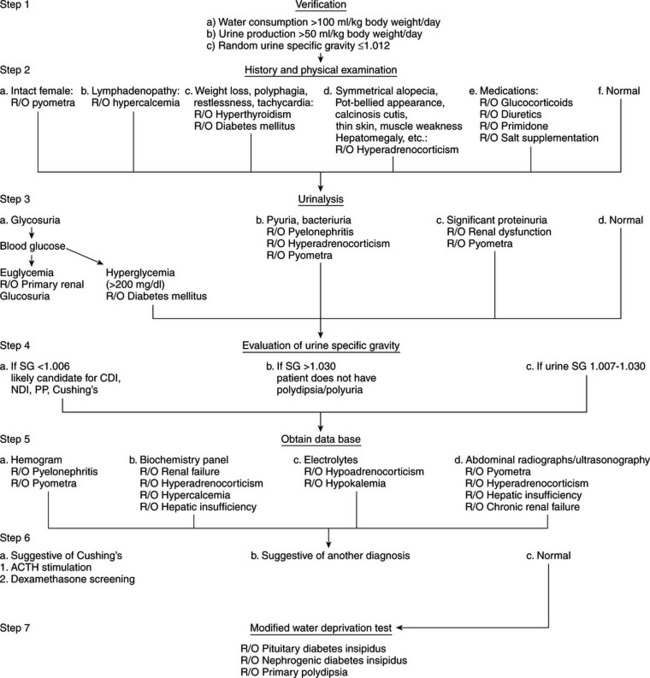
Our diagnostic approach (see Fig. 1-7) to the animal with polyuria and polydipsia is initially to rule out the more common causes. Recommended initial diagnostic studies include a CBC, urinalysis with bacterial culture of urine obtained by antepubic cystocentesis, and a serum biochemistry profile that includes liver enzymes, BUN, calcium, phosphorus, sodium, potassium, cholesterol, blood glucose, total plasma protein, and plasma albumin. A serum thyroxine (T4) concentration should be measured in older cats. Depending on the history and physical examination findings, abdominal ultrasonography may be warranted to evaluate liver, kidney, adrenal, or uterine size and to search for calcified adrenals in patients with suspected hyperadrenocorticism. Careful evaluation of the history, physical examination findings, and initial data base usually provides the diagnosis outright (e.g., diabetes mellitus, pyometra) or offers clues that allow the clinician to focus on the underlying cause (e.g., increased serum alkaline phosphatase and cholesterol in hyperadrenocorticism).
Occasionally, the physical examination and initial data base are normal in the dog or cat with polyuria and polydipsia. Viable possibilities in these dogs and cats include diabetes insipidus, psychogenic water consumption, unusual hyperadrenocorticism, renal insufficiency without azotemia, and possibly mild hepatic insufficiency. Hyperadrenocorticism, renal insufficiency, and hepatic insufficiency should be ruled out before performing tests to establish a diagnosis of diabetes insipidus or psychogenic polydipsia. Diagnostic tests to consider include tests of the pituitary adrenocortical axis, liver function tests (e.g., pre- and postprandial bile acids), urine protein: creatinine ratio, contrast imaging of the kidney. and if indicated, renal biopsy.
Careful evaluation of urine specific gravity and urine protein loss may provide clues to the underlying diagnosis (Table 1-6). For example, if the urine specific gravity measured on multiple urine samples is consistently in the isosthenuric range (1.008 to 1.015), renal insufficiency should be considered the primary differential diagnosis, especially if the BUN and serum creatinine concentration are high normal or increased (i.e., ≥25 mg/dl and ≥0.8 mg/dl, respectively) and proteinuria is present. Although isosthenuria is relatively common in dogs with hyperadrenocorticism, psychogenic water consumption, hepatic insufficiency, pyelonephritis, and partial central diabetes insipidus with concurrent water restriction, urine specific gravities tend to fluctuate above (hyperadrenocorticism, psychogenic water consumption, hepatic insufficiency, pyelonephritis) and below (hyperadrenocorticism, psychogenic water consumption, partial central diabetes insipidus) the isosthenuric range in these disorders. In contrast, if the urine specific gravity is consistently less than 1.006, renal insufficiency and pyelonephritis are ruled out and diabetes insipidus, psychogenic water consumption, and hyperadrenocorticism should be considered.
The diagnosis of diabetes insipidus and psychogenic water consumption should be based on results of the modified water deprivation test, measurement of plasma osmolality, and response to synthetic vasopressin therapy (see Confirming the Diagnosis of Diabetes Insipidus, page 21). Ideally, all realistic causes of secondary acquired NDI should be ruled out before performing tests (especially the modified water deprivation test) to diagnose diabetes insipidus and psychogenic polydipsia. The recommended initial laboratory studies not only ensure that the veterinarian is pursuing a correct diagnosis but also alert the clinician to any concomitant medical problems. A logical, systematic approach may appear cumbersome but avoids misdiagnosis. More important, problems may be avoided by not subjecting an animal to unnecessary, expensive, and potentially harmful procedures, should the presumptive diagnosis be incorrect.
ETIOLOGY OF DIABETES INSIPIDUS AND PRIMARY POLYDIPSIA
Vasopressin deficiency-central diabetes insipidus
PATHOPHYSIOLOGY.
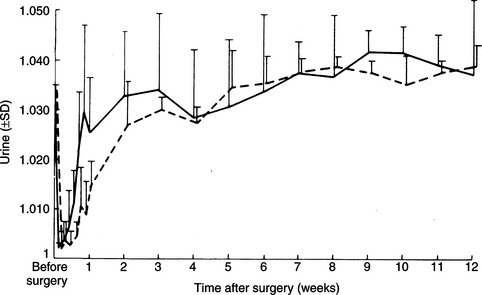
Destruction of the production sites for vasopressin–the supraoptic and paraventricular nuclei of the hypothalamus–and/or loss of the major ducts (axons) that carry AVP to the storage and release depots in the posterior pituitary (see Fig. 1-2) result in CDI. Permanent CDI requires an injury that is sufficiently high in the neurohypophyseal tract to cause bilateral neuronal degeneration in the supraoptic and paraventricular nuclei. Transection of the hypothalamic hypophyseal tract below the median eminence or removal of the posterior lobe of the pituitary usually causes transient (albeit severe) CDI and polyuria because sufficient hormone can be released from fibers ending in the median eminence and pituitary stalk to prevent occurrence of permanent diabetes insipidus (Fig. 1-8; Ramsay, 1983).
ETIOLOGY.
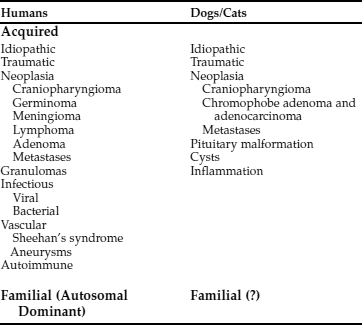
CDI may result from any condition that damages the neurohypophyseal system. Recognized causes for CDI in the dog and cat are listed in Table 1-7. Idiopathic cases of CDI are the most common, appearing at any age in any breed in either gender. Necropsies performed in dogs and cats with idiopathic CDI fail to identify an underlying reason for the AVP deficiency.
Autoimmune hypothalamitis has been suggested as a possible cause of idiopathic CDI in humans (Salvi et al, 1988). Circulating AVP cell antibodies, which bind to cell membranes of hypothalamic preparations, have been identified in some humans with CDI (Scherbaum, 1987). AVP cell antibodies have been identified prior to the development of CDI, and titers of AVP cell antibodies decline eventually to negative values with increasing duration of the disease (Bhan and O’Brien, 1982; Scherbaum et al, 1986). These patients also show a significant association with other endocrine disorders (e.g., immune thyroiditis, Addison’s disease), suggesting that, at least in some cases, polyendocrine autoimmunity may also involve the hypothalamus (see Chapter 3, page 91). A similar association between CDI and other endocrinopathies has not been identified in dogs and cats, nor have studies examining a possible immune basis for CDI been reported.
The most common identifiable causes for CDI in dogs and cats are head trauma (accidental or neurosurgical), neoplasia, and hypothalamic/pituitary malformations (e.g., cystic structures). Head trauma may cause transient or permanent CDI, depending on the viability of the cells in the supraoptic and paraventricular nuclei. Trauma-induced transection of the pituitary stalk often results in transient CDI, usually lasting 1 to 3 weeks (see Fig. 1-8; Lantz et al, 1988; Authement et al, 1989). The duration of diabetes insipidus depends on the location of the transection of the hypophyseal stalk relative to the hypothalamus. Transection at more proximal levels, close to the median eminence, is associated with a longer time for hypothalamic axons to undergo regeneration and secretion of ADH. Trauma-induced CDI should be suspected when severe polydipsia and polyuria develop within 48 hours of head trauma or when hypernatremia, hyposthenuria and hypertonic dehydration develop in a traumatized dog or cat that is being treated with intravenous fluids rather than water ad libitum (see page 29).
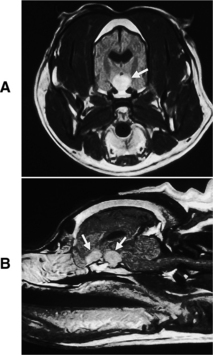
Primary intracranial tumors associated with diabetes insipidus in dogs and cats include craniopharyngioma, pituitary chromophobe adenoma, and pituitary chromophobe adenocarcinoma (Fig. 1-9; Neer and Reavis, 1983; Goossens et al, 1995; Harb et al, 1996). Tumor metastases to the hypothalamus and pituitary can also cause CDI. In humans, metastatic tumors most often spread from the lung or breast (Reeves et al, 1998). Metastatic mammary carcinoma, lymphoma, malignant melanoma, and pancreatic carcinoma have been reported to cause CDI by their presence in the pituitary gland or hypothalamus in dogs (Capen and Martin, 1983; Davenport et al, 1986). Metastatic neoplasia as a cause for CDI has not yet been reported in the cat.
< div class='tao-gold-member'>
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


