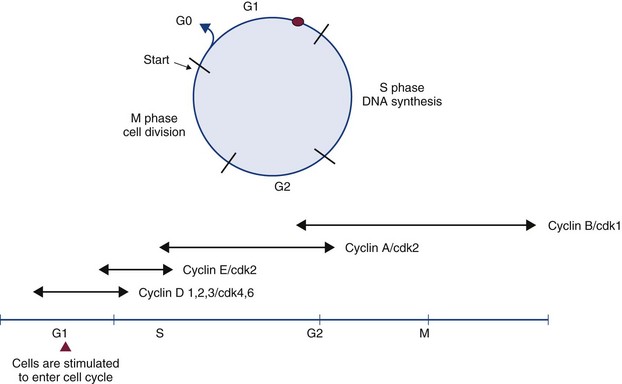
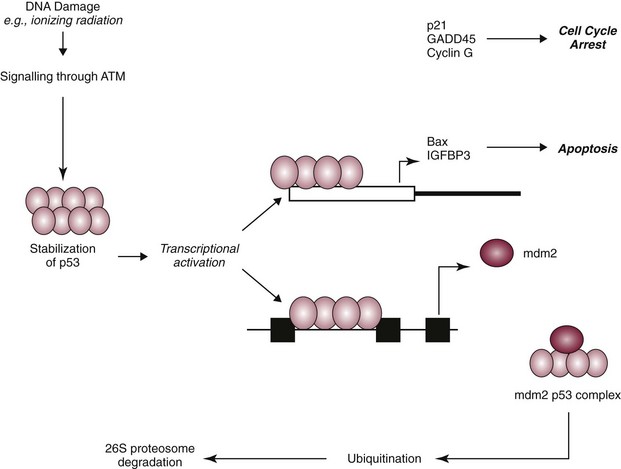
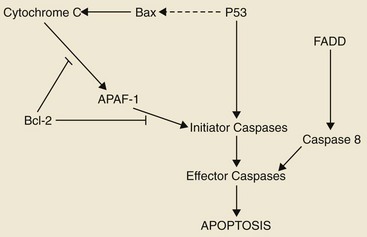
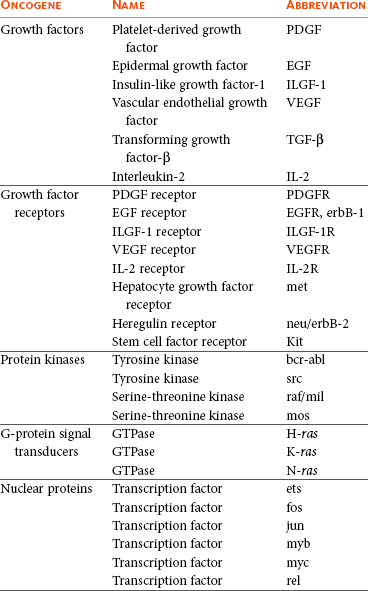
2 Cells of multicellular organisms form part of a specialized society that cooperate to promote survival of the organism. In this society, cell division, proliferation, and differentiation are strictly controlled and a balance exists between normal cell birth and the natural cell death rate.1 Derangement of these normal homeostatic mechanisms can lead to uncontrolled proliferation or loss of the ability to die, leading to a normal cell taking on a malignant phenotype. Cancer in animals is well documented throughout history but has taken on significance over the past hundred years for a number of reasons. Studies on chicken, feline, and bovine retroviruses have made significant contributions to our overall understanding of carcinogenesis through the discovery of oncogenes and tumor suppressor genes.2 Further contributions to our understanding of viral oncogenesis have come from studies of the DNA papilloma viruses in cattle and horses, complementing research into cervical cancer in women.3–10 This complementary cancer research has paved the way for the development of programs of research in comparative medicine that has benefits for both humans and the veterinary species. In this chapter we summarize the current understanding of the molecular mechanisms of cancer development and metastasis. Within an animal, all cells are subject to wear and tear, making cellular reproduction a necessity for maintenance of the individual. Reproduction of the gametes occurs by the process of meiosis, whereas reproduction of somatic cells involves two sequential phases known as mitosis and cytokinesis. Mitosis is nuclear division, and cytokinesis involves the division of the cytoplasm, the two occurring in close succession. Nuclear division is preceded by a doubling of the genetic material of the cell during a period known as interphase. As well as a copying of the chromosomes, this period is characterized by marked cellular activity in terms of RNA, protein and lipid production. The alternation between mitosis and interphase in all tissues is often referred to as the cell cycle. The phases of the cell cycle are shown in Figure 2-1. The cell cycle comprises four phases (M phase, S phase, G1, and G2) (see Figure 2-1). Nonproliferating cells are usually arrested between the M (mitosis) and S (DNA synthesis) phases and are referred to as G0 cells. The majority of cells in normal tissues are in G0. Cells are stimulated to enter the cell cycle in response to external factors including growth factors and cell adhesion. During the G1 phase of the cell cycle, cells are responsive to mitogenic signals. Once the cell cycle has traversed the restriction point (R) in the G1 phase, cell cycle transitions become autonomous. Progression through the cell cycle is mediated by the sequential activation and inactivation of a class of proteins called cyclin-dependent kinases (CDKs).11,12 CDKs consist of an inactive conserved catalytic core and are regulated at the following three levels: 1. CDK activity requires the association with regulatory subunits known as cyclins. The level of CDK remains constant throughout the cell cycle; however, the concentration of cyclins varies in a phase-specific manner during the cell cycle. The periodic synthesis and destruction of cyclins provides the primary level of cell cycle control (see Figure 2-1). 2. The activity of cyclin/CDK complexes is also regulated by phosphorylation. Activation of CDK/cyclin complexes requires phosphorylation by CDK-activating kinases (CAK); meanwhile the phosphorylation at threonine and serine residues suppresses activity. 3. CDKs are also tightly regulated by a class of inhibitory proteins known as CDK inhibitors (CDKI). CDKIs can block G1/S progression by binding CDKs/cyclin complexes and can be classified into the following two groups: The p53 response to stress may be mediated by DNA-dependent protein kinase (DNA-PK) or by the ATM kinase and leads to phosphorylation of the N terminus of p53. In normal cells, p53 is short lived; however, phosphorylated p53 is stabilized and can then function as a transcriptional regulator binding to sequences and transactivating a number of genes, including p21.13,14 p21 has a high affinity for G1 CDK/cyclin complexes and acts as a CDKI inhibiting kinase activity, thereby arresting cells in G1. By holding cells in G1, the replication of damaged DNA is prevented, and the cell’s own DNA repair machinery has the opportunity to repair damage prior to reentering the active growth cycle (Figure 2-2). The cellular levels of p53 protein are regulated by the product of another gene MDM2 (mouse double minute 2 oncogene).15 The principal role of MDM2 is to act as a negative regulator of p53 function. One mechanism for MDM2 to downregulate p53 is to target p53 for degradation. The p53 protein is maintained in normal cells as an unstable protein, and its interaction with MDM2 can target p53 for degradation via a ubiquitin proteosome pathway. MDM2 can also control p53 function by suppressing p53 transcriptional activity. MDM2 is a transcriptional target of p53, and expression is induced by the binding of p53 to an internal promoter within the mdm2 gene. MDM2 can in turn bind to a domain within the amino terminus of p53, thereby inhibiting the transcriptional activity and G1 arrest function of p53 by masking access to the transcriptional machinery (see Figure 2-2).16 In contrast to necrosis, apoptosis is a distinct type of cell death most often characterized as the “programmed” self-destruction of cells that occurs in disease states, as well as part of the normal physiologic cell turnover. Whereas necrosis is characterized by swelling of the cell and lysis, in apoptosis there is cellular and nuclear shrinkage followed by fragmentation and subsequent phagocytosis. These morphologic features of apoptosis result from a number of apoptosis effectors (i.e., caspases) and regulators (particularly the Bcl-2 protein family). The molecular mechanisms involved in apoptosis are shown in Figure 2-3.17,18 Apoptosis provides a controlled mechanism for eliminating cells that are irreversibly damaged and involves an adenosine triphosphate (ATP)-dependent activation of cellular pathways, which move calcium from the endoplasmic reticulum to the cytoplasm and activation of endonucleases. As noted previously, some of these pathways are mediated through caspases. However, a wide variety of signals can initiate an apoptotic response, including Fas ligand (CD95 or FasL) and its interaction with the Fas receptor, tumor necrosis factor (TNF) and its receptor interaction, and certain oncogenes. The Fas and TNF receptors are members of the death receptor family. These transmembrane proteins with cysteine rich extracellular domains and intracellular regions share a common structure termed the “death domain.” The proapoptotic ligands for these receptors are homotrimeric peptides that are either soluble or expressed at the surface of the adjacent cell. Ligand-induced receptor clustering promotes the binding of a soluble cytosolic adapter protein called Fas-associated death domain (FADD), which itself contains a death domain as well as a caspase binding site, to the clustered death domains of the receptors. This leads to activation of caspase 8 and downstream activation of effector caspases for apoptosis. • Benign tumors: Broadly speaking, these tumors arise in any of the tissues of the body and grow locally. Their clinical significance is the ability to cause local pressure, cause obstruction, or form a space-occupying lesion, such as a benign brain tumor. Benign tumors do not metastasize. • In-situ tumors: These are often small tumors that arise in the epithelium. Histologically, the lesion appears to contain cancer cells, but the tumor remains in the epithelial layer and does not invade the basement membrane or the supporting mesenchyme. A typical example of this is preinvasive squamous cell carcinoma (SCC) affecting the skin of cats, which is often referred to as Bowen’s disease. • Cancer: This refers to a malignant tumor that has the capacity for both local invasion and distant spread by the process of metastasis. Cancer is the phenotypic end result of a whole series of changes that may have taken a long period of time to develop. Indeed, recent studies that have sequenced the genome of pancreatic and brain tumors have identified 63 and 60 genetic alterations on average in each cancer, respectively. From this large list of genetic alterations, there are a small number of commonly mutated genes that are “drivers” of the cancer phenotype.19,20 The application of a cancer-producing agent (carcinogen) to tissues does not lead to the immediate production of a cancer cell. After the initiation step produced by the agent, there follows a period of tumor promotion. This promotion may be caused by the same initiating agent or by other substances, such as normal growth promoters or hormones. The initiating step is a rapid step and affects the genetic material of the cell. If the cell does not repair this damage, then promoting factors may progress the cell toward a malignant phenotype. In contrast to initiation, progression may be a very slow process and may not even manifest in the lifetime of the animal. Each stage of multistep carcinogenesis reflects genetic changes in the cell with a selection advantage that drives the progression toward a highly malignant cell. The age-dependent incidence of cancer suggests a requirement for between four and seven rate-limiting, stochastic events to produce the malignant phenotype.21 The RNA tumor viruses (retroviruses) provided the first evidence that genetic factors play a role in the development of cancer. The initial observation came in 1910 when Rous demonstrated that a filterable agent (later classified as a retrovirus and termed avian leukosis virus) was capable of producing lymphoid tumors in chickens.22 Retroviruses have three core genes (gag, pol, and env) and an additional gene that gives the virus the ability to transform cells. Retroviral sequences that are responsible for transforming properties are called viral oncogenes (v-onc). The names of these genes are derived from the tumors in which they were first described (e.g., v-ras from rat sarcoma virus). Viral oncogenes were subsequently shown to have cellular homologues called cellular oncogenes (c-onc). Later, the term proto-oncogene was used to describe cellular oncogenes that do not have transforming potential to form tumors in their native state but can be altered to lead to malignancy.23 Most proto-oncogenes are key genes involved in the control of cell growth and proliferation and their roles are complex. For simplicity, their sites and modes of action in the normal cell can be divided as follows (Table 2-1): The advent of recombinant DNA technology has allowed scientists to unravel a number of mechanisms by which the normal products of proto-oncogenes can be disrupted to produce uncontrolled cell division. The conversion of a proto-oncogene to an oncogene is a result of somatic events in the genetic material of the target tissue. The activated allele of the oncogene dominates the wild-type allele and results in a dominant gain of function. This means that only one allele needs to be affected to obtain phenotypic change; this is in contrast to tumor suppressor genes in which both alleles have to be lost for phenotypic change. The mechanisms of oncogene activation are outlined in the following list and are shown in Figure 2-4.23–30 • Chromosomal translocation. Where proto-oncogenes are translocated within the genome (i.e., from one chromosome to another), their function can be greatly altered. The classic example in human medicine is the chromosomal breakpoint that produces the Philadelphia chromosome found in chronic myelogenous leukemia (CML). This involves the translocation of the c-abl oncogene on chromosome 9 to a gene on chromosome 22 (bcr). The point at which two genes come together is referred to as a chromosomal breakpoint (or translocation breakpoint). The BCR/ABL hybrid gene produces a novel transcript whose protein product has elevated tyrosine kinase activity and can contribute to uncontrolled cellular proliferation. Transgenic mice for this chimeric gene develop lymphoblastic leukemia and lymphoma. Since this gene product is directly linked to CML formation, it is a logical target for tyrosine kinase inhibitors in the treatment of chronic myeloid leukemia in humans (CML). • Gene amplification. Quantitation of gene copy number is possible by a number of molecular techniques, including comparative genomic hybridization, genotyping arrays, and Southern hybridization. Amplification of oncogenes can occur in a number of tumor types and has been demonstrated in childhood neuroblastoma, where the myc proto-oncogene (nuclear transcription factor) is amplified up to 300 times. Gene amplification is possibly the most common mechanism of proto-oncogene activation. A further example is the MDM2 proto-oncogene, which has been identified in dogs and horses, and has recently shown to be amplified in a proportion of canine soft tissue sarcomas.31 • Point mutations. These are single base changes in the DNA sequence of proto-oncogenes leading to the production of abnormal proteins. Point mutations can arise through the actions of ionizing radiation, chemical carcinogens, or errors in DNA replication and repair. A mutation in a proto-oncogene or the transcriptional machinery that controls its expression may disrupt homeostasis and result in sustained proliferation signals or a failure to respond to negative feedback signals. A classical example is the Ras proto-oncogene in which point mutations are a consistent finding in a number of human tumors. K-ras mutations have also been identified in canine lung tumors. Mutations in the Erb-B (epidermal growth factor receptor) gene have been shown in a number of human cancers to lead to ligand-independent activation. Erb-2 mutations have been identified in canine mammary tumors. • Viral insertions. The discovery of oncogenes was a direct result of studies on tumor-causing viruses. In some circumstances, proto-oncogene function can be damaged by the insertion of viral elements. Occasionally, novel retroviruses are isolated from leukemias or sarcomas in animals that have been viremic with a leukemia virus for some time. These viruses induce tumors very rapidly when inoculated into members of the species of origin and are referred to as acutely transforming oncoviruses. The prototype of the acutely transforming virus is the Rous sarcoma virus (RSV) isolated from a fowl in 1911. Subsequently, many more have been isolated from animals infected with avian, feline, murine, or simian oncoviruses. These viruses are generated by a rare recombinatorial event between the leukemia virus with which the animal was originally infected and a cellular proto-oncogene. In this, part of the viral genome is deleted and replaced with the cellular oncogene. The virus then becomes acutely transforming because this oncogene is now under the transcriptional control of very efficient viral promoters. This then allows infection of a cell and insertion of this continuously expressed oncogene into the cellular genome leading toward rapid progression and malignancy. Evidence suggests that these acutely transforming viruses are not transmitted naturally, but all events occur in the individual animal. Because the virus has itself lost some of its own genetic material, it is defective for replication. However, they are spread throughout an animal by the provision of help from the normal leukemia virus, which provides the missing proteins in co-infected cells. Retinoblastoma occurs in two forms, a sporadic form and an inherited form (accounting for 40% of cases).32 In the inherited form, the mode of inheritance is autosomal dominant and about half the children are affected by the condition. Knudson’s model required the retinoblastoma tumor cells (in either sporadic or inherited forms) to acquire two separate genetic changes in the DNA before tumor development. The first or predisposing event could be inherited through the germ line (familial retinoblastoma) or it could arise de novo in somatic cells (sporadic form). The second event occurred in somatic cells. Thus, in sporadic retinoblastoma, both events arose in the retinal cells. However, in familial retinoblastoma, the individual had already inherited one mutant gene and only required a second hit in the remaining normal gene in somatic cells. The p53 protein has been described as the guardian of the genome by virtue of its ability to push cells into arrest or apoptosis, depending on the degree of DNA damage. Thus the p53 tumor suppressor gene plays an important role in cell cycle progression, regulation of gene expression, and the cellular response mechanisms to DNA damage. Under normal physiologic conditions, wild-type p53 can bind specific DNA sequences and regulate transcription of a number of genes involved in cell cycle progression and apoptotic pathways including p21waf1/cip1 and bax (see Figure 2-2). The p53-mediated mechanisms are responsible for tumor suppression and prevent accumulation of potentially oncogenic mutations and genomic instability. Failure by p53 to activate such cellular functions may ultimately result in abnormal uncontrolled cell growth leading to tumorigenic transformation.33–36 The homologs of p53 and MDM2 have both been identified in domestic animal species and a number of studies indicate that this gene also has a central role in the progression of veterinary cancers.37–41 The advances in our understanding of normal cell biology and the processes that lead to malignancy have increased dramatically over the past 30 years. The last decade has shown us that transformation of a normal cell into a malignant cell requires very few molecular, biochemical, and cellular changes that can be considered as acquired capabilities.42,43 Further, despite the wide diversity of cancer types, these acquired capabilities appear to be common to all types of cancer. An optimistic view of increasing simplicity in cancer biology is further endorsed by the fact that all normal cells, irrespective of origin and phenotype, carry similar molecular machineries that regulate cell proliferation, differentiation, aging, and cell death. Cancer cells have defects in regulatory circuits that govern cellular proliferation and homeostasis and survival. A model has been proposed that suggests that the vast array of genetic abnormalities associated with cancer are a manifestation of eight alterations in cellular physiology that collectively contribute to malignant growth. First proposed in 2000 and updated in 2011, these “hallmarks” of cancer constitute an organizing principle for rationalizing the complexities of cancer and are underpinned by two overarching themes: genome instability and chronic inflammation.42,43 The eight acquired characteristics can be summarized under the following headings (see Figure 1-3): • Insensitivity to antigrowth signals • Evasion of programmed cell death (apoptosis) • Limitless replicative potential • Reprogramming energy metabolism The cancer cell can acquire this capability in the following ways: • They may produce growth factor ligands themselves. • They may induce stromal cells to produce such ligands. • There may be an increase in receptor concentration on the cell surface that leads to receptor homodimerization or heterodimerization, making the cell hyperresponsive to ligands. • There may be structural alterations in the receptor to support ligand-independent firing. • Constitutive activation of the signaling pathway (downstream of the receptor). A good example of this is the constitutive activation of the PI3-AKT pathway, through mutations in the catalytic subunit of the PI3 kinase. • Disruptions in negative-feedback mechanisms that attenuate proliferative signaling, such as the following: • Mutations in the Ras gene compromise the Ras GTPase activity, which acts as a negative-feedback mechanism to ensure the effects of Ras are only transitory. • PTEN is a tumor suppressor protein that counteracts PI3 signaling. PTEN loss has a similar effect to constitutive PI3 activation and promotes tumorigenesis. Most cellular programs that negatively regulate cell growth and proliferation depend on the actions of tumor suppressor genes. At the basic level, most of the antiproliferative signals are funneled through the Rb protein and its close relatives. Disruption of Rb allows cell proliferation and renders the cell insensitive to antiproliferative signals such as that provided by the well-characterized transforming growth factor-β (TGF-β).32 The Rb protein integrates signals from diverse extracellular and intracellular sources and can control cell cycle progression. The other major tumor suppressor is p53, which integrates intracellular signals and can promote either cell cycle arrest or apoptosis (depending on the degree of cellular stress or damage). However, the effects of p53 expression are highly context dependent. Loss of Rb or p53 is associated with the malignant phenotype through the cell’s ability to evade antigrowth signals. • Another characteristic of the cancer cell is the evasion of contact inhibition. Cell-to-cell contact in most normal cells results in an inhibitory signal against further cell proliferation. The role of this mechanism in vivo has been thought to be to maintain tissue homeostasis. In cell culture, contact inhibition is abrogated in cancer cell monolayers, leading to their indefinite expansion. NF2 and LKB1 genes are considered tumor suppressor genes that are involved in this process and loss of these genes in vivo may promote loss of contact inhibition that contributes to the progression of cancers. • Although TGF-β has antiproliferative effects in cancer, it is now, however, appreciated that the TGF-β pathway can be corrupted in the later stages of malignancy and can contribute to cancer progression. In this late effect, TGF-β is found to activate a cellular program termed epithelial-to-mesenchymal transition (EMT) that promotes invasion and metastasis (see later).
Tumor Biology and Metastasis
Normal Cell Division
The Cell Cycle
Cellular Responses to DNA Damage
p53 Functions as a Genomic Guardian
Cell Death
From Normal Cell to Cancer Cell
Multistep Carcinogenesis
Oncogenes
Nuclear Proteins and Transcription Factors
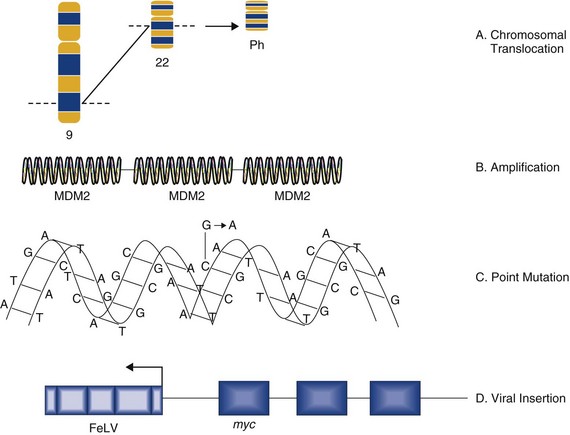
Mechanisms by which Oncogenes Become Activated
Tumor Suppressor Genes
Retinoblastoma Forms the First Clues to the Existence of Tumor Suppressor Genes
The p53 Tumor Suppressor Gene
Cancer Arises through Multiple Molecular Mechanisms
The Hallmarks of Cancer
Self-Sufficiency in Growth Signals
Insensitivity to Antigrowth Signals or Evading Growth Suppressors
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Tumor Biology and Metastasis