Chapter 14 Therapy of Cardiovascular Diseases
Cardiovascular Physiology as it Pertains to Cardiovascular Drugs
Membrane Ion Movements and the Action Potential
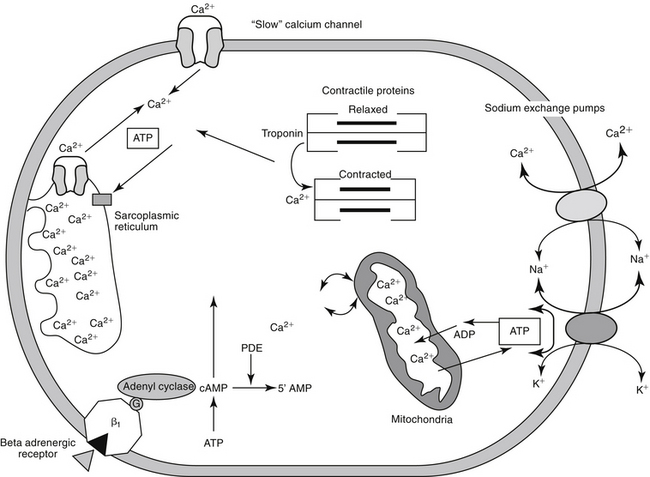
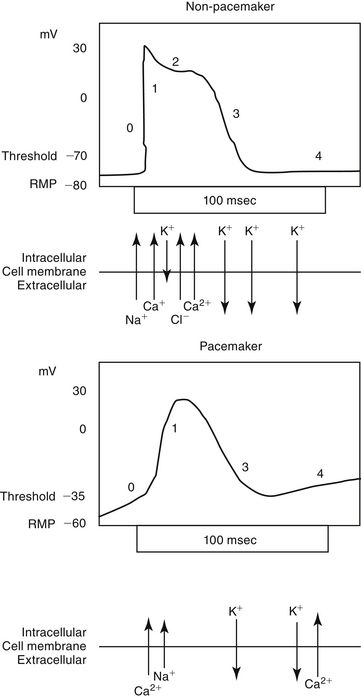
Predicting the nuances of pharmacodynamic responses to cardiac drugs depends, in part, on understanding the electrophysiology of myocardial cells. Selected mechanisms of ion movement into the cell are demonstrated in Figure 14-1. The action potential duration (APD) of myocardial cells is long compared with that of nerves, reflecting the well-orchestrated coordination of multiple ion channels and associated transport proteins.1 The magnitude and direction of the ion flow, and thus current, depends on both transmembrane voltage and ion concentration gradients. Two types of myocardial cells will be discussed: those capable of automaticity (i.e., spontaneous depolarization), exemplified by cells that normally serve as pacemakers (e.g., sinoatrial [SA] node) and those cells not normally capable of automaticity (e.g., atrial and ventricular myocardial cells). Although overlap exists, the electrophysiology of each differs from one another (Figure 14-2).
Nondepolarizing cells
Movement of ions through the channels follows both concentration and electrical (i.e., electrochemical) gradients. Ion flow is also influenced by energy-dependent pumps that actively and selectively direct ion movement against the electrochemical gradients. A “leak current” allows a constant, albeit smaller, ion flux. At rest the status of all channels influencing the membrane potential is static, save for a specific (“inward rectifier”) channel that is permeable only to potassium, allowing it to efflux down an electrochemical gradient. This efflux is countered by a Na+,K+-ATPase pump, which exchanges 3Na+ for 2K+, thus maintaining the intracellular concentration necessary for the gradient. Accordingly, potassium has the greatest influence on the RMP, and very slight changes in extracellular potassium can influence potassium flux, the RMP, and the cardiac cell cycle.
Myocardial sodium channels are the primary gatekeepers of the action potential in nonpacemaker cells (see Figure 14-1). They are voltage gated and closed at rest. Membrane depolarization in nonpacemaker cells (see Figure 14-2) increases selective permeability in sodium channels. The concentration gradient causes an initial sodium influx, stimulating more voltage-gated sodium channels to open, thus perpetuating a self-regenerating action potential (phase 0, depolarization). Sodium channels inactivate when depolarization is complete and must go through a resting (recovery) period before they can be reactivated. Membrane permeability reestablishes itself largely in response to potassium ion efflux, and the membrane potential begins to decline back toward the RMP (phase 1, early repolarization). As the membrane potential continues to repolarize (phase 3), the ability of sodium channels to reactivate increases, until the RMP is once again reached (phase 4). Because sodium influx and sodium channel recovery is rapid, sodium channels are referred to as “fast,” in contrast to “slow” calcium channels, which take longer to recover. Drugs with greater affinity for inactivated sodium channels generally are more able to affect the refractory period and the APD compared with those that target either resting or open channels. Sodium influx during phase 0 depolarization influences other ion channels and their ion movements.
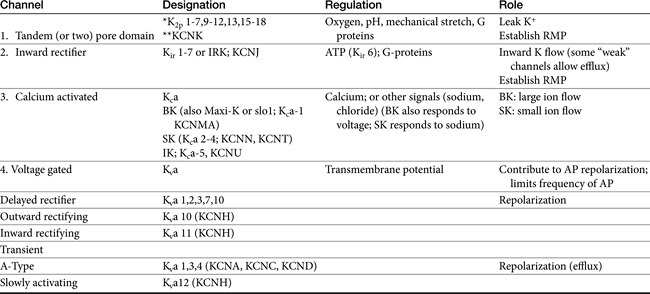
Among the ions most influenced by sodium flux in nonpacemaker cells are potassium channels, of which multiple types, exist, varying in tissue and intracellular location, and control (Table 14-1). Inward rectifier potassium channels are among the channels that determine the RMP. Depolarization causes conformational changes in transient potassium channels, which rapidly inactivate, and delayed rectifier potassium channels, which include both slow and rapid inactivators (each voltage gated). Efflux of potassium through these channels causes the membrane to begin to repolarize. Initially, the outward potassium current is balanced by an inward depolarizing current of calcium through L-type channels, causing the plateau, or phase 2, of repolarization in nonpacemaker (but not pacemaker) cells. Increased activity of delayed rectifier channels increases potassium efflux, which, when coupled with closing calcium channels, results in late repolarization (phase 3; see Figure 14-2). Slower delayed rectifier potassium channels remain open to ensure net outward potassium efflux such that the membrane potential becomes more negative, causing rapid delayed rectifier and inwardly rectifying potassium channels to open. The delayed rectifier K+ channels close when the membrane potential is restored to about -80 to -85 mV, but the inwardly rectifying potassium channels remain open throughout phase 4, thus maintaining the RMP. Because potassium channels generally remain open as long as the membrane is not at its resting potential (i.e., is in some state of depolarization), potassium channels also influence the duration of the action potential. Thus the influence of potassium channels in nondepolarizing cells includes the RMP and the rate of repolarization. In pacemaker cells (discussed in greater depth later in this chapter), potassium channels influence the RMP, the rate of repolarization, and the initial rate of phase 4. Consequently, potassium and drugs that target potassium channels potentially influence all cardiac (e.g., depolarizing and nondepolarizing) tissues.
Calcium also plays a major role in both nondepolarizing and depolarizing myocardial cells. The extracellular to intracellular calcium ratio of myocardial cells approximates 10, generating a calcium ion flow down both a concentration and electrical gradient. In contrast to the vasculature, in which channel movement is predominantly receptor mediated, calcium flux in the myocardium is predominantly voltage dependent. At least three types of voltage-gated calcium channels exist in the cardiovascular system, with differences reflecting conductance and sensitivity to voltage: T, N, and L types.2 The N-type (neuronal) calcium channels are located predominantly in neural tissues and markedly differ in responsiveness compared with L and T calcium channels. The best characterized of the calcium channels are of the L-type (long-lasting, large), and they are the predominant influencing channel during the states of the cardiac action in which the membrane potential is positive. Despite being voltage gated, calcium L-channels contain receptors (similar to those in the vasculature) that influence calcium channel flow and are subject to drug-induced blockade. In contrast to L-type channels, T-type (transient, tiny) channels are more active at negative potentials and are the predominant channel of influence at rest. As such, whereas L- channels are activated at high voltages, T-channels are activated at low voltages.
In nonpacemaker cells, in response to sodium influx, extracellular calcium enters the cell through L-type channels as repolarization begins. The influx is sufficient to stimulate release of intracellular calcium from the sarcoplasmic reticulum and other Ca2+ stores (e.g., mitochondria). This (initial sodium and subsequent) calcium ion flow links the action potential to excitation–contraction coupling in nondepolarizing cells.3 The duration of contraction is determined by the rate of intracellular calcium removal from actin and myosin sites in the cytosol by either resequestration into the sarcolemma (or other stores) or efflux from the cell. Efflux is accomplished by at least two pumps, both of which exchange sodium for calcium. The Na+/Ca2+ ATP-ase pump exchanges 1 Na+ for 3 Ca2+ but has only a minor influence on phase 2 of repolarization. The plateau of phase 2 repolarization in nondepolarizing cells is primarily influenced (sustained) by a balance between inward movement of calcium through L-type channels and outward movement of K+ through the slow delayed rectifier potassium channels.4
As with sodium channels, calcium channels exist in resting (closed, but responsive), open, or inactivated (unresponsive) states, and recovery must occur before the resting (responsive) state is achieved. Because both current movement and recovery of calcium channels are slower compared with those of sodium channels, the term slow channels is used to refer to ion movement through calcium channels. Hormones, neurotransmitters, and inorganic ions influence calcium channels. As with sodium channels, those drugs with a greater affinity for inactivated calcium channels are more effective.
Refractory Periods
Myocardial cells are neither excitable nor responsive to additional stimuli during the early and intermediate phases of the action potential cycle. Further, they are only partially responsive if stimulated before complete repolarization has occurred—that is, as the RMP returns to normal. Refractory periods (RPs) describe the period of time (or proportion of the APD) in which cells are nonresponsive. During the absolute refractory period (ARP), cells are totally nonresponsive. In contrast, cells may be partially (effective; ERP) or relatively (RRP) refractory to stimuli, with the state dependent on the number of open (i.e., recovered from inactivation) sodium channels. The ARP occurs when sodium channels are closed. Tissues will not respond to any stimuli, no matter how strong. The ERP is the shortest interval that can occur before a premature impulse can propagate a response.5 It includes the ARP plus a shorter period following the ARP that reflects the opening of some sodium channels but not enough to transmit an impulse. The RRP follows the ERP and represents a state in which a sufficient number of sodium channels are open such that a very strong stimulus might be propagated. The refractory periods are protective in that they limit the rate at which myocardial tissues can respond to impulses, thus ensuring sufficient time for cardiac filling and ejection to occur before the next contraction occurs. Proper direction of impulse propagation is also facilitated. Because sodium channels during these periods are refractory to reopening, unilateral (one direction only) conduction is ensured along a myocardial fiber, precluding the premature regeneration of an action potential and inappropriate coordination between excitation–contraction coupling. Loss of unilateral conduction is a contributing factor to re-entrant or circus rhythms.
Myocardial Contractility
Contraction of both cardiac and smooth muscle depends on Ca2+. The inotropic state of the muscle reflects the relationship between resting fiber length and peak isometric tension. The myocardium develops force for contraction and thus the strength to pump blood by forming cross-bridges between actin and (tropo) myosin myofilaments in cardiac muscle. The amount of force that the muscle can generate depends on the number of cross-bridges that form when myosin engages actin. Energy in the form of adenosine triphosphate (ATP) causes a sliding motion between the proteins of the myofilaments and cardiac muscle to shorten and develop force. The interaction between proteins in the myofilament is regulated by troponin, which is found at regular intervals on the tropomyosin fibers. Troponin is formed from three subunit proteins: T binds to tropomysin, I inhibits the actin-binding site on tropomyosin, and C binds to calcium.4 Calcium binding of troponin C forces a conformational change in the troponin complex, causing troponin I to move away from tropomyosin, thus allowing cross-bridging between actin and myosin. The force that develops as actin and myosin interact depends on both the affinity and amount of calcium binding to troponin. The amount is regulated by the concentration of intracellular calcium. It is only as intracellular calcium is removed that troponin I moves back into position; thus contraction will continue until all intracellular calcium is removed.
Multiple mechanisms influence myocardial intracellular (cytosolic) calcium (see Figure 14-1). Extracellular calcium can enter through two sources: movement through (slow) electrogenic or voltage-gated calcium L channels embedded in T-tubules, which ensures that calcium is delivered in close proximity to the sacroplasmic reticulum, and movement though Na+-Ca2+ ATP channels, which is dependent on cell membrane ATPase.3,6 Cytosolic calcium flow is modulated primarily by β-adrenergic receptors; increased cyclic adenosine monophosphate (cAMP) activates protein kinase, which in turn increases calcium movement through the L-channels.4 Opening of slow calcium channels in response to depolarization causes intracellular calcium to rise rapidly, stimulating subsequent release from intracellular storage sites (sarcoplasmic reticulum and, to a lesser degree, mitochondria). The contracted myocardial muscle relaxes as intracellular calcium concentration falls as a result of resequestration into the sarcoplasmic reticulum and efflux from the cell, both of which are energy (ATP) dependent. Disorders of lusitropy (i.e., disorders of diastolic relaxation) occur if intracellular calcium does not decrease. The velocity and extent of cardiac muscular contraction are regulated by sarcomere length.4 Length (stretch) reflects preload, or the transmural filling pressure. An optimal stretch maximizes the relationship between actin and myosin filaments, allowing more Ca2+-activated cross-bridges and more forceful contractions. In the normal cat and dog, the upper limit of filling pressure in the left ventricle stretches the sarcomere to the length that generates peak tension during contraction. With sustained systolic overloading of the heart, however, the ideal sarcomere stretch is exceeded, cross-bridging decreases and myocardial contractility declines. Abnormalities of the excitation–contraction coupling mechanism contribute to the pathogenesis of cardiomyopathies and chronic hemodynamic overloading.
The most important factor regulating myocardial contractility is stimulation of cardiac sympathetic nerves; cAMP serves as the secondary messenger, altering intracellular calcium flux and myocardial contractility. Myocardial cAMP is produced by adenylyl cyclase, which in turn is regulated by either stimulation or inhibition of adenine or guanine nucleotide proteins. Many cell surface receptors interact with proteins that regulate adenylyl cyclase. An increase in intracellular cAMP causes phosphorylation of proteins that increase calcium influx through the “slow” calcium channels, and the release, reaccumulation, and storage of calcium in the sarcoplasmic reticulum. Cyclic AMP is degraded by several phosphodiesterases (PDEs) isoenzymes, each of which has been associated with specific pharmacodynamic actions. At least 11 isoforms have been named. Inhibition of these enzymes causes the same effect as an increase in either adenylyl or guanylyl cyclase and thus cAMP or cGMP, respectively (see Figure 14-1). The pharmacodynamic response varies with tissue site: PDE II is located in smooth muscle of the urinary bladder detrusor muscle; PDE III in the heart, systemic vascular smooth muscle, and platelets (cAMP); PDE IV in bronchial smooth muscle and pulmonary circulation (cAMP); PDE V in the smooth muscle of the corpus cavernosum, visceral smooth muscle, skeletal muscle, platelets, kidney, lung, cerebellum, and pancreas (cGMP); and PDE VI in the retina (responsible for transduction).
Adrenergic Receptors
The adrenergic nervous system has a major physiologic role in modulating normal myocardial inotropic and chronotropic states and the time-variable tension that develops as ventricles contract. Both the myocardium and peripheral vasculature are innervated with sympathetic nerve terminals. Under normal conditions norepinephrine released from nerve endings in the heart acts as the primary regulator. Circulating catecholamines released from the adrenal gland play a less important role in normal conditions, but their influence increases as myocardial failure progresses. Molecular cloning techniques have identified nine subclasses of receptors: alpha (α) 1 (three subclasses); α 2 (three subclasses); and β 1, 2, and 3.7 Adrenergic receptors are linked to different G protein–coupled receptors, which differentially influence secondary messenger systems (sometimes the same one). Beta receptors are linked to adenylyl cyclase through Gs proteins. Beta agonists regulate cell processes by increasing cAMP, thus influencing downstream effects through cAMP-dependent protein kinases (see the discussion of smooth muscle). In contrast, α receptors are linked to Gi proteins, which oppose the actions of Gs proteins, thus decreasing cAMP.
In the normal heart, stimulation of the sympathoadrenal system is the primary method by which the heart adjusts to transient changes in workload. The myocardium possesses predominantly β receptors whereas vascular smooth muscle is rich in α receptors. Both β receptors and α receptors are subdivided into two types. Beta-1 receptors predominate in the myocardium, increasing inotropy (strength of contraction) and chronotropy (rate of impulse generation) (see Figure 14-1). Myocardial effects of adrenergic receptors are achieved through increased magnitude of the calcium current, slowed channel inactivation, and increased magnitude of K+ and Cl– repolarizing currents. Pacemaker current and thus sinus rate increase.5 Beta-2 receptors (and recently described β-3 receptors) are also located in the heart, but their function is not clear. Disease affects the state of receptors. Continued stimulation of adrenergic receptors, such as that which accompanies diseases states (e.g., congestive heart failure) or long-term adrenergic therapy, results in a dampening or desensitization of response to receptor stimulation. Desensitization reflects internalization and destruction of cell surface receptors. For example, β-1 receptors decrease up to 75% (β-2 receptors are spared) in human patients with congestive heart failure, leading to a compensatory increase in sympathetic signal outflow, which likely contributes to the pathophysiology (see the discussion of myocardial remodeling).7 The function of myocardial β-3 receptors is not clear, but they may provide feedback inhibition of contractility; an imbalance in myocardial disease may contribute to the pathophysiology of myocardial disease.8
In vascular smooth muscle, α-2 receptors mediate vasodilation (Figure 14-3). Most α activity in the cardiovascular system is mediated by way of α-1 receptors. Effects include contraction of vascular (and nonvascular) smooth muscle. A-2 receptors inhibit neurotransmitter release but also mediate vascular contraction (as do α-1 receptors). Subtypes 1 and 2 of either α or β receptors can be selectively pharmacologically stimulated (agonists) or inhibited (antagonists) to manage cardiovascular disease.
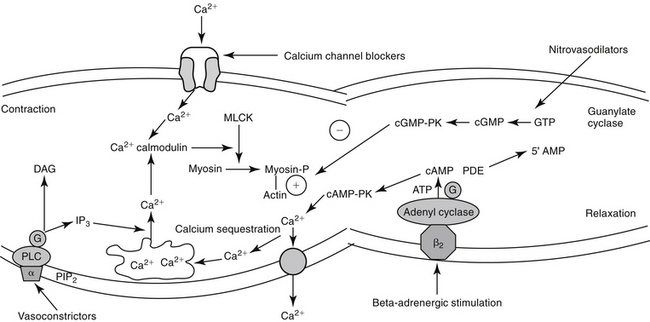
Smooth Muscle of the Vasculature
Myocardial oxygen demand is directly related to heart rate, myocardial wall tension, and the inotropic state of the myocardium.3,9 Myocardial wall tension is determined by the size (diameter) of the ventricle and intraventricular pressure. Thus tension is affected by preload (end-diastolic volume and stretch) and afterload (aortic blood pressure). Drugs that decrease systemic arterial pressure through dilation of arterioles decrease left ventricular afterload. Following the path of least resistance, a larger volume of blood will be ejected from the ventricular chamber into systemic circulation, thus diverting blood from the pulmonary vasculature. Left ventricular filling (preload) and thus myocardial wall size and tension will decrease, as will myocardial oxygen demand. An advantage of preload or afterload is that the decrease in cardiac work occurs without affecting myocardial contraction.
KEY POINT 14-9
A number of mediators act to cause arterial constriction and thus increase peripheral resistance.
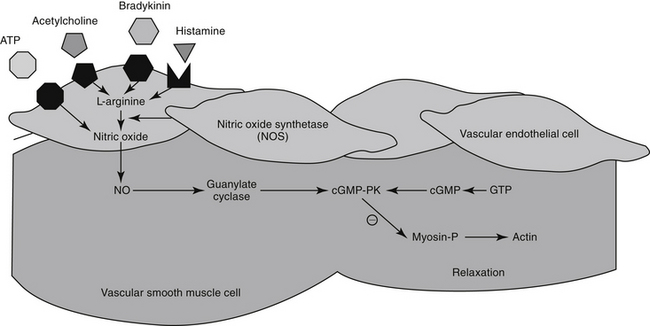
The excitation–contraction coupling in vascular smooth muscle depends on calcium influx, which enters the cell through either voltage-sensitive (electrogenic) signals associated with depolarization or, more commonly, through receptor-operated Ca2+ channels. Intracellular calcium also is released from the sarcoplasmic reticulum in response to membrane phosphatidylinositol hydrolysis and formation of the secondary messenger inositol triphosphate.2 Intracellular calcium interacts with calmodulin, activating myosin light-chain kinase (MLCK) to phosphorylate myosin light chain. Cross-bridging between myosin and actin causes smooth muscle to contract. Cyclic AMP decreases both MLCK and intracellular calcium, causing relaxation of vascular smooth muscle. As with cAMP, the secondary intracellular messenger cGMP causes vascular smooth muscle relaxation, although the mechanism is different (see Figures 14-1 and 14-3). Endothelium-derived relaxing factor (EDRF; chemically related to nitric oxide) and endothelium-derived constricting factor (EDCF) are among the vasoactive substances released by the endothelial cell that control the hemodynamics of the cardiovascular system. Intracellular response to EDRF (or nitric oxide) is probably signaled by cGMP. Other mediators of vascular smooth muscle response include, but are not limited to, prostacyclin, histamine, and acetylcholine. Mediators released from the endothelial cell generally act locally on vascular smooth muscle (Figure 14-4); an exception might include mediators released from the pulmonary vasculature, which may be sufficient to modulate a systemic response.
Systolic and diastolic pressures are, respectively, the upper and lower limits of the oscillations around mean arterial pressure. The mean arterial pressure is the arterial pressure over time and is defined as the diastolic pressure plus one third of the pulse pressure. Arterial blood pressure is the product of cardiac output (determined by stroke volume and heart rate) and total peripheral resistance. Total peripheral resistance is the sum of resistance in all vascular beds. It is also affeccted by aortic impedance (resistance to flow) and diastolic arterial pressure, which in turn is determined by the sympathetic nervous system, the renin–angiotensin–aldosterone system and arginine vasopressin system, vascular (extracellular fluid) volume, and aldosterone or other volume active hormones.43
The Renin–Angiotensin–Aldosterone System
The renin–angiotensin–aldosterone system (RAAS) plays an important role in regulating blood volume, arterial pressure, and cardiac and vascular function (Figure 14-5).10 An additional but critically important role in cardiac repair and remodeling has recently emerged. RAAS regulation of arterial pressure is accomplished through constriction of resistance vessels, mediated by several mechanisms. Included are direct stimulation of AGII receptors and indirect stimulation through facilitation of norepinephrine. Vasopressin also is a potent mediator of peripheral vasoconstriction by way of V-1 receptors. Its increase reflects either increased release from sympathetic nerve terminal endings or decreased reuptake.11
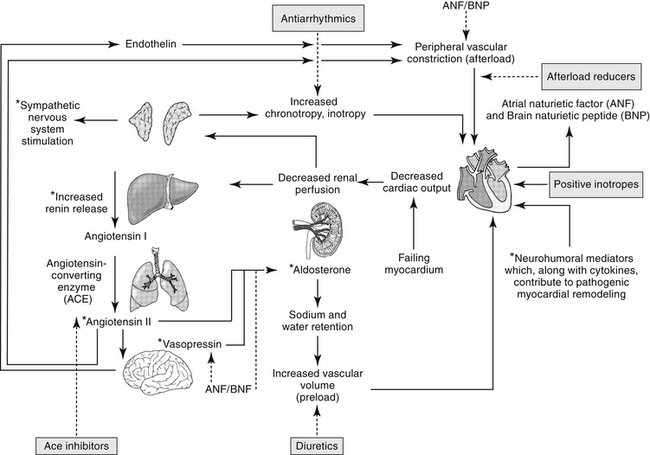
ACE is a kinase II metallopeptidase enzyme bound to the membrane of a variety of cells, but particularly endothelium, epithelium, neuroepithelium, and brain cells.12 Organs respond to both systemic and local renin, with local response influenced by local concentrations of ACE and angiotensin receptors. Concentrations of ACE differ among tissues, with that in the renal tubular brush border the greatest (300- and 10-fold higher than the left ventricle and lung, respectively). Thus, although the kidney is the most important site of renin release, it also is a target of the RAAS, responding to both systemic and urinary renin. Renal AGII concentrations exceed circulating AGII more than 1000-fold, causing renal vasoconstriction and sodium retention. Degradation of AGII yields angiotensin III (AgIII), which has 40% of the pressor and 100% of the aldosterone effects of AGII.
Through AGII, RAAS modulates responses to low sodium intake and provides for long-term control of renal function, body fluid volumes, and arterial pressure. In the healthy canine kidney, response to AGII results in increase in both preglomerular and postglomerular vascular resistance, although the predominant effect is on the efferent rather than afferent arteriole. Renal blood flow consequently decreases, but glomerular capillary pressure increases.12 AGII regulates body fluid content by directly stimulating thirst centers in the brain; adrenal release of aldosterone, which mediates increased renal sodium and fluid retention; and posterior pituitary release of vasopressin (antidiuretic hormone, ADH), a component of the arginine vasopressin system (AVP). Vasopressin increases renal fluid retention through V2 receptors in renal tubular cells. In addition to its vascular effects, AGII released from endothelial cells also facilitates cardiac hypertrophy and vascular hypertrophy. Notably, production of inflammatory cytokines is increased from both normal and abnormal (damaged) myocardial tissue, contributing to the negative sequelae of cardiac remodeling.13 AGII is prothrombotic and may induce cardiac muscle hypertrophy.
Renal vascular response to AGII is mediated by AGII receptors (ARs), a transmembrane G-coupled protein receptor consisting of several subtypes that vary in location, numbers, affinity for AGII, and secondary messenger systems. The most well known of the subtypes, AR-1, preferentially binds AGII and AGIII, mediating the classic angiotensin RAAS responses on blood pressure and water and electrolyte balance. Included are water, sodium intake, renal sodium retention, secretion of vasopressin and aldosterone, and cell growth/proliferation. In addition to AR-1, AR-2 receptors also bind AII and AIII.14–16 However, AR-2 receptor density is greatest in the brain, including areas involved with fluid and electrolyte regulation and balance, arterial pressure, cognition, behavior, and locomotion. Concentrations of AR-2 also are high in steroid-producing glands, including the adrenal glands and ovaries.16 Because of the central location of AR-2, AGII and AGIII can modulate many body functions and responses. Expression of AR-2 is particularly high during fetal development, but it persists in the adult brain, supporting a role in neuronal function. In the brain, AR-2 appears to oppose the traditional RAAS effects of AR-1 on drinking behavior and vasopressin secretion. Other effects mediated by AR-2 receptors are regulation of cell proliferation, apoptosis, and cellular differentiation.16 Again, AR-2 appears to attenuate AR-1 mediated apoptosis, pressor, and chronotropic effects. Like AR-1 receptors, several secondary systems appear to signal AR-2 effects, including the mitogen-activated protein kinase (MAPK) and nitric oxide/cGMP pathways.16
Regulation of RAAS effects through angiotensin receptors is complex, possibly involving feedback inhibition pathways, responses that vary with duration of exposure, and systems that may oppose one another. Likewise, pharmacologic management of heart disease through manipulation of AR receptors may be complex.15 Disease is likely to contribute to variability in response to AGII and its modulating drugs. For example, in the failing heart the expression of AR-1 decreases, whereas that of AR-2 does not change or increases. Both receptors are associated with effects that initially, in moderation, might benefit the patient, but with progression become detrimental. Not surprisingly, a link has been described between AR and β-adrenergic receptors. Both AR-1 and β receptors interact with G proteins, with AR-1 through activation of phospholipase C and β-receptors through activation of adenylyl cyclase. Diamerization of the two receptors has been described in vitro and occurs in vivo.17 This integration will further complicate pharmacologic manipulation.
Mechanisms other than ACE modulate formation of AGII. Opposing effects of ACE are regulated in part by AR, but other body systems also modulate the influence of ACE. For example, ACE also inhibits breakdown of bradykinin. Bradykinin consequently increases, resulting in vasodilation and naturiesis.18 The vasodilatory effects of bradykinin are mediated in the vascular endothelium through arachidonic acid derivatives, nitric oxide, and endothelium-derived hyperpolarizing factor in the vascular endothelium. The mechanism of natriuresis is not clear.12 Bradykinin also has beneficial effects on cardiac remodeling, which helps oppose the negative sequelae of AGII.
The Role of Nitric Oxide
For decades, researchers have attempted to identify a factor released from endothelial cells, referred to as EDRF, which is responsible for mediating a number of stimuli causing vasodilation. Ultimately, nitric oxide (NO) was recognized to be the smallest and most basic mediator of vascular response.19 Released as a gas (and thus often mistaken for nitrous oxide [N2O], or “laughing gas”), it is synthesized in response to NO synthetase (NOS) enzymes from L-arginine and oxygen or by sequential reduction of inorganic nitrate (see Figure 14-4). However, as a free radical, NO is very reactive and unstable and interacts with oxygen on exposure to air to form the pollutant nitrogen dioxide (NO2). Two major classes and three isoforms of NOS have been identified.19-21 Constitutive NOS (cNOS or NOS-1) is continuously produced and includes two isoforms synthesized either by vascular endothelial cells (eNOS) or neurons (nNOS). Constitutive NOS, which is calcium dependent, tends to mediate cell responses through cellular receptors. Responses of cNOS include that mediated by vascular mediators such as acetylcholine, norepinephrine, histamine, and substance P. Not surprisingly, response is rapid. Inducible NOS (iNOS or NOS-2) is produced as needed by inflammatory cells (e.g., macrophages, neutrophils, and Kuppfer cells), generally after exposure to cytokines (e.g., tumor necrosis factor or interleukins) or bacterial lipopolysaccharides. Production of NO from iNOS requires new protein synthesis and is characterized by a delay of several (2 to 4) hours.
Regardless of origin, NO causes its effect by diffusing across cellular membranes to intracellular targets. Cytosolic cGMP is the major intracellular messenger (see Figure 14-4) causing physiologic response to NO. Responses include dilation of blood vessels, inhibition of thrombogenesis, cytotoxic responses, and neuronal signaling. However, because NO contains an unpaired electron in its outer orbit, it is a free radical. As such, it can contribute to the formation of other radicals while simultaneously scavenging oxygen radicals. The half-life of NO is so short that studies involving NO generally are based on its oxidation end products nitrates and nitrites.20 However, despite its very short half-life, NO has many important and complex actions in the body. Under basal conditions, peripheral vasoconstriction is locally relieved by intermittent cNOS-induced NO in response to sheer stress and endothelial cell receptor stimulation. Inflammation and immune signals also induce NO release by way of iNOS. NO inhibits platelet aggregation and adhesion, contributing to antithrombogenic mechanisms in the vascular endothelium. NO may ameliorate the detrimental effects of norepinephrine on the growth of cardiac myocytes and fibroblasts, suggesting that increased NO bioavailability may prevent or reverse remodeling in patients that have experienced heart failure.22 Modulation of inflammation varies, however, with cell type and the source of NO production (i.e., iNOS versus cNOS). Although targeting NO production through drug therapy may appear to be a reasonable approach to the treatment of a variety of cardiovascular disorders, the complex nature of its release and the events leading to its release currently preclude predictable and safe modulation. It is likely, however, that selective modulation of NO ultimately will provide a therapeutic approach to many disorders.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


