Chapter 5 Therapeutic Drug Monitoring
Therapeutic drug monitoring (TDM) is a tool for guiding the design of an effective and safe regimen for drug therapy in the individual patient. Monitoring can be used to confirm a plasma drug concentration (PDC) that is above or below the therapeutic range, thus minimizing the time that elapses before corrective measures can be implemented for the patient.1–3 Knowledge of the principles of drug disposition (see Chapter 1) and the factors that determine these principles in the individual patient (see Chapter 2) facilitate an understanding of the use of and need for TDM.
Fixed dosing regimens are designed to generate PDCs within a therapeutic range—that is, achieve the desired effect without producing toxicity. Dosing regimens are based on the patient’s clinical response to the drug. Therapeutic success is most likely to occur if doses are based on scientific studies performed in the target species intended to receive the drug for the intended reason. Marked interindividual variability has, however, been confirmed for many drugs4,5 owing to physiologic (e.g., species, breed, age,6 gender), pathologic (e.g., renal, hepatic, cardiac diseases)7–10 or pharmacologic (i.e., drug interaction)5,7,11,12 effects. Prudent clinicians modify dosing regimens when possible to compensate for the impact of some of these factors on drug disposition. However, the combined effects of these factors are often unpredictable. A trial-and-error approach to dose modification may be successful but is most appropriate when response to the drug can be easily measured. Examples include “to effect” drugs such as gas inhalants and ultrashort thiobarbiturate anesthetics, rapidly acting anticonvulsants such as diazepam, and lidocaine for the treatment of ventricular arrhythmias. The trial-and-error approach also might be reasonable for illnesses that are not serious or do not require immediate resolution and for drugs characterized by large therapeutic windows, that are generally safe at high doses, However, trial-and-error modification can be inefficient and potentially dangerous when the drug response cannot be easily measured, the drug is characterized by a narrow margin of safety, or the patient’s life is threatened.
Drugs and Indications
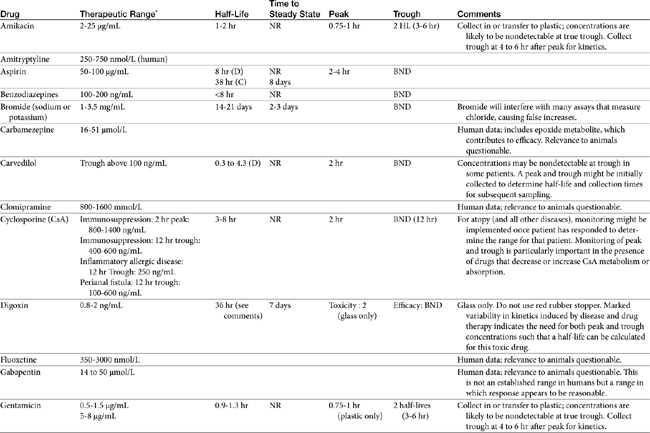
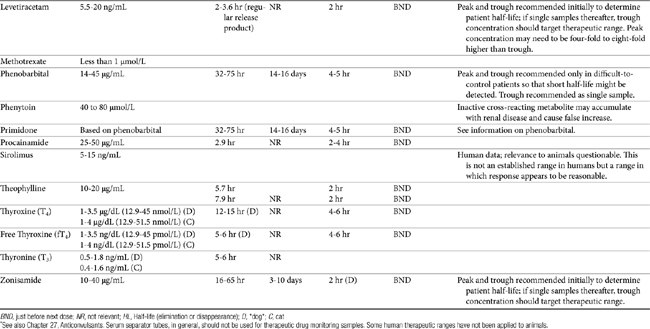
TDM is not indicated for all drugs; rather, it is indicated when patient health is at risk (Box 5-1). Not all drugs can be monitored by TDM; certain criteria must be met (Box 5-2). 13 Patient response to the drug must correlate with PDC. Drugs whose metabolites are active (e.g., diazepam) or for which one of two enantiomers compose a large proportion of the desired pharmacologic response generally cannot be monitored effectively by measuring the parent drug.14 Rather, all active metabolites, the parent drug, or both or the pharmacologically active enantiomer should be measured. The drug must be measurable at concentrations within the targeted therapeutic range in a relatively small sample size, and analytic methods must be available to detect the drug rapidly, precisely and accurately in the target species.15 Methods must be specific for the drug of interest and able to differentiate it from other compounds. Prior to choosing a laboratory for monitoring purposes, the laboratory also should be queried regarding quality-control procedures to ensure that they are followed and assays used in animals have been validated for that species. Ideally, the laboratory will participate in some type of external validation program. Attention should be given to how the sample collected from the patient is handled. The cost of the analytic method must be reasonable. Drugs that meet these criteria and for which TDM has proved useful in veterinary medicine include, but are not limited to, selected anticonvulsants (phenobarbital, bromide, selected benzodiazepines zonisamide and levetiracetum), antimicrobials (e.g., aminoglycosides-gentamicin, and amikacin); cardioactive drugs (digoxin, procainamide, lidocaine, and quinidine); theophylline, and cyclosporine (Table 5-1).
Box 5-1 Drugs for Which TDM Is Most Useful
Box 5-2 When to Implement Monitoring
Monitoring may be most effective if either or both minimum (Cmin) and maximum (Cmax) ranges have been established for the drug in the species and for the disease being treated.4 However, the importance of the therapeutic range should not be overestimated. Although not “normals,” therapeutic ranges of drugs also are population statistics, based on the PDC at which most patients (e.g., 95%) with the targeted disease might be expected to respond. However, although the therapeutic range offers a reasonable target for most animals, exactly where in the range the individual patient will respond is not known. It is this patient-specific concentration that is identified through monitoring—that is, monitoring establishes the therapeutic range for the individual patient. As such, it is indicated to establish the baseline response in a patient that has adequately responded to therapy. Note that some animals will respond at concentrations below or above an established therapeutic range. However, dosing regimens need not necessarily be increased or decreased, respectively, for those patients unless the patient is put at risk for subtherapeutic failure or toxicity, respectively.
Therapeutic ranges do offer a target for drug therapy and are ideally based on well-controlled clinical trials in the target species. However, most recommended therapeutic ranges in animals have been extrapolated from those determined in humans. Although these ranges have proven useful, none-the-less, studies confirming the applicability of these ranges to animals are warranted. Determination of these ranges can be facilitated if adequate patient information accompanies a patient sample submitted for monitoring (see Box 5-1). Procainamide is an example of a drug for which recommended therapeutic ranges might differ between dogs and humans because of pharmacokinetic differences. Dogs do not produce the acetylated active metabolite as efficiently as humans; therefore procainamide concentrations should be higher in dogs than in humans. Primidone (rarely used currently) is an example in cats: its efficacy in dogs depends on conversion to phenobarbital, which does not happen to a significant degree in cats. Bromide offers another example of pharmacodynamic differences: whereas concentrations above 1.5 mg/mL might be considered toxic in people, they are in the low- to mid-therapeutic range in epileptic dogs. The therapeutic range for a drug also varies with the therapeutic intent (e.g., cyclosporine and perianal fistulas versus immunosuppression).
As with clinical pathology reference ranges, the range for a specific laboratory also may vary with the methodology, and specifically whether or not metabolites are detected by the methods. As such, the laboratory should be specifically queried regarding its therapeutic range for a particular assay. Assays that are based on antibodies (e.g., enzyme-linked immunosorbent assay [ELISA], radioimmunoassay [RIA], polarized immunofluorescence [PIFA]) may detect both the parent compound and those metabolites most chemically similar to it. Monoclonal antibody–based immunoassays are less likely to detect metabolites than polyclonal antibody-based assays but nonetheless, may not be able to discriminate among very subtle changes in drug chemistry induced by metabolism. Therapeutic ranges for antibody-based assays generally are higher than those that detect only the parent compound. Which assay is preferred depends on the activity of the metabolites: if the metabolite is active, immunoassays may more accurately predict response compared with assays based on parent compound only. Cyclosporine, benzodiazepines, procainamide, behavior modifying drugs, and beta blockers are examples of drugs that might have active metabolites.
Implementation
Number and Timing of Samples: One Versus Two, Peak Versus Trough
For drugs with a long half-life compared to the dosing interval, because drug concentrations will not fluctuate significantly during the interval, variability in drug concentrations may reflect instrument variability, rather than drug disposition. For such drugs (e.g., bromide and, in most patients, phenobarbital or zonisamide), the timing of sample collection probably is not important and a single sample collected at any time is sufficient. However, a trough sample just before the next dose is almost always the lowest concentration that will occur with each dose, and as such is generally recommended if a minimum concentration must be maintained (e.g., anticonvulsants). In general, response to therapy and TDM should not be performed unless maintenance dosing has been sufficiently long for PDC to reach “steady state” (3 to 5 drug half-lives). An exception includes situations in which a loading dose has been administered (see below). If a loading dose is not given, the drug will accumulate (see Chapter 1) as steady state is reached (Figure 5-1). The time to steady state varies among drugs and for some drugs is quite variable among animals (see Table 5-1).
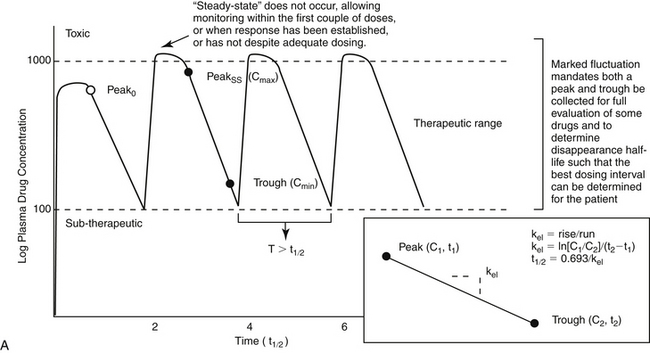
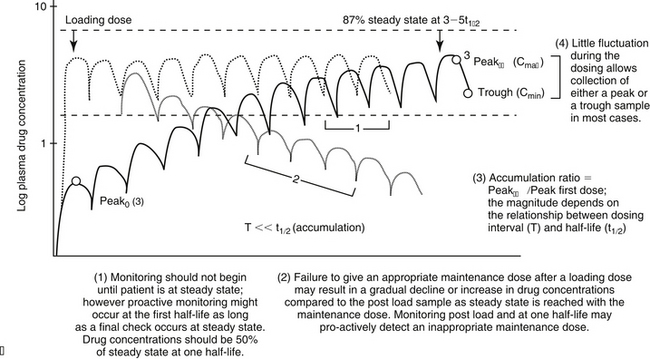
For some drugs, the elimination half-life changes across time in the patient. The decision to collect both a peak and trough for such drugs may depend on the elimination half-life, which in turn, can be documented as short only by collecting both a peak and a trough sample. This is exemplified by phenobarbital, for which the elimination half-life may initially be longer than the dosing interval (i.e., more than 48 hours) but after induction (i.e., several months into therapy) may be much shorter (i.e., less than 12 hours) in the same patient (Case Study 5-1). Digoxin provides a good example of the risk associated with collecting only a single sample when assessing efficacy. Digoxin is characterized by a half-life that ranges from less than 12 hours, thus allowing concentrations to become subtherapeutic during a 12-hr dosing interval, to more than 36 hours (particularly in patients with renal disease), which is likely to lead to drug accumulation (Case Study 5-2). The half-life can change again if the patient responds to therapy for cardiac failure. If toxicity is suspected, a single sample collected at the time that clinical signs of toxicity appear can confirm toxicity. Neither toxicity nor efficacy can be confirmed throughout the dosing interval, however, unless two samples (peak and trough) are collected (Case Study 5-3).
If a kinetic profile of a patient is the reason for TDM, at least two samples must be collected to establish a PDC-versus-time relationship (Case Study 5-5). For such drugs, the samples preferably are collected at the peak and trough times. Regardless of the route of drug administration, with two data points, elimination (disappearance) half-life can be calculated. However, a more comprehensive kinetic profile can be built from the same two data points for a patient receiving an intravenous dose: volume of distribution (Vd) and clearance can then be estimated in addition to drug elimination half-life.
Loading doses warrant a special note. Loading is implemented with the goal of achieving steady state concentrations immediately. The advantage is to avoid the delay that otherwise will occur as steady state is gradually reached. The disadvantage is that the body will not have time to accommodate to side effects. As such, loading should be limited to situations in which failed response might be life-threatening. Although the design of the loading dose may be successful in achieving target steady state concentrations (see calculations below), the patient is not yet at steady state and will not be until the same dosing regimen has been implemented for 3 to 5 half-lives. Thus, as the patient transitions from the loading to the maintenance dose, the time to steady state begins again. If the maintenance dose fails to maintain what the loading dose achieved, then drug concentrations will gradually decline or increase until steady state is achieved (Figure 5-1, B). Because the majority of the change will occur in the first half-life of maintenance dosing, clinical signs of failure or toxicity may be more likely to occur during this initial half-life. Proactive monitoring after the loading dose has been absorbed and again at the first half-life will allow the clinician to assess the effectiveness of the maintenance dose. However, both samples must be collected for the assessment. Collecting a sample one half-life after the loading dose without a postload sample is minimally informative. For example, when using a loading dose for bromide, TDM should be performed three times. The first time to monitor is after oral absorption of the last of the loading doses to establish what the loading dose accomplished (i.e., day 6). The second time is at one drug half-life later (e.g., at 21 days for bromide) to ensure that the maintenance dose is able to maintain concentrations achieved by loading. Collection of this second sample at the one drug half-life point is recommended because most of the change in drug concentrations that will occur if the maintenance dose is not correct will occur during the first half-life. If the second sample (collected at one drug half-life) does not approximate the first (collected immediately after the loading doses), the maintenance dose can be modified at this time rather than wait for steady state, with the risk of therapeutic failure or toxicity. The third time to monitor bromide when using a loading dose is at steady state (e.g., 3 months) to establish a new baseline.
The timing of sample collection also must take into account the pharmacodynamic response of some drugs. For example, many antimicrobials (e.g., aminoglycosides) are characterized by a half-life that is less than 2 hours (e.g., amikacin in dogs) but are given at much longer dosing intervals (e.g., 24 hours). The peak sample is important for determination of efficacy of this concentration-dependent drug. Because 12 half-lives will have elapsed before the next dose, less than 2% of the original dose will remain in the body at true trough concentrations, and concentrations are not likely to be detectable at true trough (i.e., 24 hrs after dosing). Although the absence of detectable drug may be sufficient information if safety is the reason for monitoring an aminoglycoside, kinetics (e.g., half-life) cannot be determined. Therefore collection of both a 2-hour peak and and a sample two to four elimination half-lives later will allow calculation of a half-life and extrapolation of concentrations at trough (Case Study 5-5). Cyclosporine is another example: if administered every 24 to 48 hours, because its half-life in normal dogs approximates 4 to 5 hours, little drug will be detected at 24 to 48 hours.
Specific timing of peak PDC collection is more difficult to determine accurately than that of trough concentrations. Peak PDC should be determined after drug absorption and distribution are complete (see Chapter 1). Route of drug administration can influence the time at which peak PDCs occur, which varies among drugs. For orally administered drugs, absorption is slower (1 to 2 hours), and distribution is often complete by the time peak PDCs have been achieved. The absorption rate can, however, vary widely due to factors such



