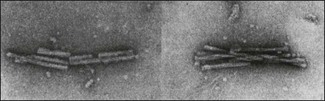
Chapter 67 Several neurodegenerative diseases of animals and man belong to a group known as transmissible spongiform encephalopathies (TSEs), which are viewed as part of a larger group of neurodegenerative diseases including Alzheimer disease and Parkinson disease in humans referred to as ‘protein-folding diseases’. These diseases share a number of characteristic features including appearance late in life, neuronal loss and the accumulation of deposits of misfolded protein aggregates in the CNS (Soto & Estrada 2008). Transmissible spongiform encephalopathies are caused by prions (Prusiner 1982) and have several common features including: prolonged incubation period, progressive course, invariably fatal outcome, similar neuropathological changes, accumulation of an abnormally folded host-derived prion protein. Prions are defined as proteinaceous, infectious particles that resist inactivation by procedures which modify nucleic acids. They appear to be composed exclusively of a modified isoform of a host-derived protein, prion protein (PrP), and are non-immunogenic. The normal or cellular form of the prion protein referred to as PrPC is composed of about 208 amino acids and occurs on the surface of many types of cells, particularly neurons and lymphocytes. The function of PrPC is still largely unknown. The disease-associated or scrapie form of the prion protein, in reference to the prion disease of sheep, is denoted PrPSc and accumulates as aggregates called scrapie-associated fibrils to form deposits in the brain and lymphatic tissue of affected individuals (Fig. 67.1). Both PrPC and PrPSc are encoded by a single copy chromosomal gene. Differences between PrPC and the corresponding PrPSc reside in their tertiary structure and not in amino acid composition. Much of the polypeptide chain of PrPC is coiled up into structures termed α-helices. Conversion of PrPC into PrPSc involves a post-translational change resulting in a largely β-sheet structure. This structural transition produces profound changes in physico-chemical properties including loss of solubility in non-denaturing detergents and a dramatic increase in resistance to proteases. Figure 67.1 Electron micrograph of extensively purified scrapie-associated fibrils (SAF), composed of prion protein, PrP. Negatively stained with uranyl formate. Bar represents 100 nm. Reprinted with permission: Veterinary Virology Third Edition (1999). Murphy et al., Academic Press. Page 575. Substantial advances have been made in understanding the molecular features of TSE agents (Silveira et al. 2004). A number of theoretical models of PrPSc formation have been proposed, in particular the heterodimer model (Prusiner 1998) and the nucleation (seed)-dependent polymerization model (Jarrett & Lansbury 1993). These two models can overlap, for example auto-catalysis and the formation of a metastable PrPC folding intermediate may be features of both. It has been shown in vitro that conditions of mild acidification and reduction, similar to those of endosomes or lysosomes, promote the unfolding and rearrangement of PrPC resulting in a monomeric form rich in β-sheets (Jackson et al. 1999, Zou & Cashman 2002). Co-factors appear to play an important role in prion propagation and infectivity (Ma 2012). The β-sheet-rich form of PrP (β-PrP) may convert back to the α-conformation or may form a stable ‘seed’ that promotes the abnormal folding and polymerization of PrPC. If several β-PrP molecules bind together to form a multimer they lock irreversibly in the β-conformation of PrPSc. During the process fission occurs whereby the long PrPSc polymers fragment resulting in an increase in the number of effective nuclei to direct further conversion of PrPC. The conditions required for the conversion process may occur during recycling of PrPC. Most membrane glycoproteins destined for degradation or recycling are transported to lysosomes via endosomes. Studies indicate that PrPSc is formed in caveolae-like domains before they fuse with endosomes and that PrPSc accumulates in cytoplasmic vesicles particularly lysosomes (Prusiner et al. 1999). Prion replication may be initiated: • Following exposure to a ‘seed’ of PrPSc as seen in acquired disease cases. • As a result of a rare stochastic event whereby there is spontaneous conversion of PrPC to β-PrP as occurs in sporadic prion disease cases. • As a result of a mutation in the PrP gene which gives rise to PrPC pre-disposed to form β-PrP as seen in inherited prion disease cases. Prions are classified into two types, mammalian and fungal. The agents of spongiform encephalopathies are distinguished by their disease association and host species. The primary amino acid sequence and thus the species of a particular prion is determined by the sequence of the chromosomal PrP gene of the animal in which it last replicated. The resistance of some animals to the inoculated prions from another species is termed the ‘species barrier’. It is primarily governed by differences in the amino acid sequence and conformation. According to the conformational selection model, ease of transmission between species is determined by the degree of overlap or compatability between the PrPSc types permitted by the host PrPC and the introduced PrPSc (Collinge & Clarke 2007). The inoculation of a prion species from one host into another host species results in prolonged incubation times during the first passage. Subsequent passage in this second host species results in a shortening of the incubation time. The ‘species barrier’ has been used to explain why humans have not contracted scrapie from sheep. Strains of prions that ‘breed true’, particularly scrapie strains, have been described based on the results of bioassays in mice. The determination of a strain is achieved using the incubation period and mortality pattern in inbred mice of particular known genotypes, the distribution and extent of spongiform lesions and prion protein plaques in the brain (‘lesion profile’) and the titre of infectivity in the brain. Although the primary structure of PrP is an important determinant of the tertiary structure of PrPC, PrPSc is believed to act as a template for the conversion of PrPC into nascent PrPSc, thereby determining the tertiary structure of nascent PrPSc molecules. Prion diversity is thought to be enciphered in the conformation and glycosylation pattern of PrPSc such that prion strains represent different conformers of PrPSc. The existence of strains has been cited as evidence of the involvement of nucleic acid and the ‘protein only’ theory of prions is still debated (Chesebro 1998). Prions are stable over a very wide pH range and remarkably resistant to physical or chemical inactivation. Underestimation of the resistance of these agents has resulted in a number of tragic human cases as well as the dissemination of scrapie agent to 18,000 sheep in a louping ill vaccine prepared from formolized suspensions of brain, spinal cord and spleen (Greig 1950). In fact the treatment of prions with alcohols or aldehydes that fix proteins helps to protect these agents from inactivation and to enhance their thermostability. Extensive research has been carried out into physical methods of inactivating the agents of BSE and scrapie due to the importance of disposing of carcases of affected animals and the importance of meat and bone meal as a source of infection for ruminants. Autoclaving at temperatures of 132 to 138°C does not guarantee inactivation. In fact, under certain conditions the effectiveness of autoclaving actually declines as the temperature is increased. The most effective methods under worst-case conditions are hypochlorite solutions containing 2% available chlorine or heated 2N sodium hydroxide (Taylor 2000). The inclusion of a formic acid step in formaldehyde fixation of brain tissue has been shown to dramatically reduce the infectivity of scrapie, BSE and CJD agents without significantly affecting the quality of histological sections. The effectiveness of formic acid is most likely due to its solubilizing effect on proteins. Prion diseases are significantly affected by the genome of the host. In the case of the rare human diseases fatal familial insomnia and Gerstmann–Straussler–Scheinker disease (GSS) the condition is inherited in an autosomal dominant manner. Three different manifestations of prion diseases are described (Prusiner 1997) and best exemplified by Creutzfeldt–Jakob disease (CJD) in man which presents as an infectious, sporadic and inherited disorder: • Slow infection. Kuru is a slowly progressive disease transmitted by ritualistic cannibalism formerly practised by the Fore people, inhabiting the Eastern Highlands of Papua New Guinea. A number of cases of CJD (iatrogenic CJD) have been associated with the use of human cadaver tissues or extracts such as corneal transplants and human growth hormone. Several prion diseases of veterinary importance involving slow infection with very long incubation periods are described (Table 67.1). Certain polymorphisms of the PrP gene in sheep are strongly associated with the incidence of scrapie to the extent that some researchers have inferred from breeding studies that scrapie is an exclusively genetic disease. However, Australia and New Zealand are free of scrapie despite the presence of animals with scrapie-associated PrP alleles (Hunter et al. 1997). The epizootic of bovine spongiform encephalopathy (BSE) in Great Britain is believed to have resulted from changes in rendering practices permitting the preservation of prion proteins, possibly derived from scrapie-infected sheep, in meat and bone meal (Wilesmith 1993). New variant CJD cases in man were first reported in Great Britain in the mid 1990s and are believed to result from the ingestion of beef containing the agent of BSE. Table 67.1 Transmissible spongiform encephalopathies of veterinary significance
Prions (proteinaceous infectious agents)
Clinical infections
Disease
Host
Significance of infection
Scrapie
Sheep, goats
Exact mode of transmission unknown, probably maternal transmission in pre or early post natal life. Long incubation period and progressive, nervous disease of adult sheep. Pruritis is a common feature. Recognized in Europe since 1700s. Worldwide distribution, except Australia and New Zealand
Bovine spongiform encephalopathy
Cattle
First described in 1986 in Great Britain at start of major epizootic. Principally confined to Great Britain with much lower incidence in many other European countries. Controlled by effective exclusion of ruminant-derived protein from diet of cattle. Novel form of CJD described in man in 1996 associated with the epizootic
Chronic wasting disease
Mule deer, white-tailed deer and elk
Present in farmed and wild deer populations in North America. Mode of transmission unclear but probably through environmental contamination with infected saliva and faeces. Affects adult animals. Characterized by wasting, behavioural changes and nervous signs
Exotic ungulate encephalopathy
Greater kudu, nyala, oryx and other exotic ungulates in zoological collections
Infection associated with feeding of meat and bone meal derived from BSE-affected animals
Transmissible mink encephalopathy
Mink
Sporadic disease of ranched mink. Infection associated with feeding of meat and offal from prion-infected sheep and cattle carcases. Control readily achieved by exclusion of foodstuffs derived from sheep, cattle or other mink
Feline spongiform encephalopathy
Cats
Infection associated with feeding of tissues derived from BSE-affected animals. Long incubation period with clinical course of several months characterized by nervous signs. Majority of cases reported in Great Britain. Control readily achieved by exclusion of contaminated bovine tissue from cat food ![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Prions (proteinaceous infectious agents)
Only gold members can continue reading. Log In or Register to continue
