Prions
Agents of Transmissible Spongiform Encephalopathies
Abstract
This chapter describes the properties of prions and features of prion diseases, also known as transmissible spongiform encephalopathies, in animals and humans.
Keywords
Prions; transmissible spongiform encephalopathy; mad cow disease; variant Creutzfeldt–Jakob disease; scrapie; chronic wasting disease
The term “transmissible spongiform encephalopathy” is used for several neurodegenerative diseases: scrapie of sheep and goats, bovine spongiform encephalopathy, feline spongiform encephalopathy, transmissible mink encephalopathy, chronic wasting disease of cervids, and four human diseases: kuru, Creutzfeldt–Jakob disease (including variant Creutzfeldt–Jakob disease (vCJD)), Gerstmann–Sträussler–Scheinker syndrome, and fatal familial insomnia. These uniformly fatal diseases are caused by prions—ie, “infectious proteins” or “rogue proteins.” The name prion is an acronym derived from the words protein and infectious. In each of these diseases, the characteristic lesion is spongiform degeneration with activation and proliferation of astrocytes and microglia in the brain and spinal cord.
The prototypic prion disease, scrapie, was first recognized in England in 1732, and a report from 1750 clearly describes scrapie as an infectious and consistently fatal disease of sheep. The name reflects the characteristic scratching observed in diseased animals. Scrapie is enzootic in sheep in all countries except Australia and New Zealand. In 1963, Dr. William Hadlow, a veterinary pathologist, first proposed that the brain lesions observed in humans with kuru were similar to those of scrapie in sheep, and that kuru might be transmissible following a long incubation period. Kuru, a fatal neurological disease, occurred only in the Fore tribe in the New Guinea highlands, where ritualistic cannibalism was practiced on deceased relatives. Hadlow’s idea about kuruled to the discovery by Dr. Carleton Gajdusek that kuru could be transmitted to chimpanzees, causing a disease indistinguishable from the human counterpart and similar to scrapie. The importance of this discovery became clear when it was shown that more common human diseases such as Creutzfeldt–Jakob disease, and other animal diseases, such as chronic wasting disease of deer and elk, are also transmissible.
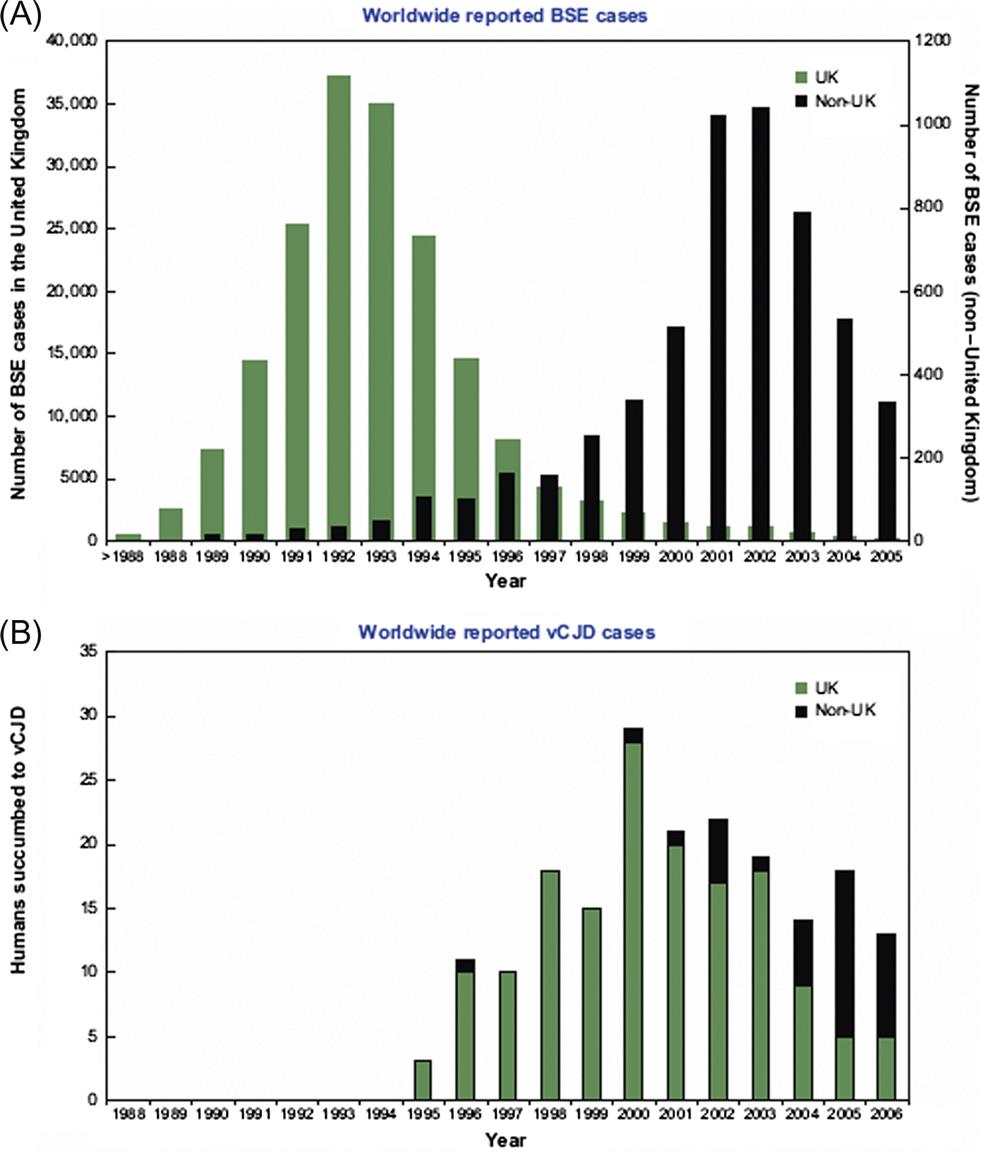
Bovine spongiform encephalopathy (“mad cow disease”) was first detected in 1986 in the United Kingdom. Epidemiological observations suggest that the cattle disease originated in the early 1980s and became established in cattle through recycling of rendered bovine meat and bone meal in the ruminant food chain. As more and more diseased cattle were slaughtered and rendered to produce meat-and-bone meal, a massive, multiple-point-source epizootic followed. Export of meat-and-bone meal from the United Kingdom introduced the disease into many other European countries, and to Canada. The disease also was introduced into zoo animals and domestic and exotic cats in the United Kingdom through the same source. In 1996, the British government first announced that humans had probably become infected with the bovine spongiform encephalopathy prion through exposure to cattle products. By June 2014, the number of human cases of what is now called “variant Creutzfeldt–Jakob disease” (vCJD) had risen to 174 in the United Kingdom and 52 in other countries, although many of these latter cases had resided in the United Kingdom.
Epidemiologic, pathologic, and molecular studies have strengthened the causative association between the bovine prion and the human disease. At the heart of this association were research breakthroughs on the nature of prions and the mechanisms of their pathogenicity; in 1997, Dr. Stanley Pruisner was awarded the Nobel Prize in Medicine for his discovery of novel infectious proteins and their exceptional mechanism of amplification. Halting the practice of recycling bovine meat and bone meal led to a rapid decline in bovine spongiform encephalopathy cases in the United Kingdom over 7 years (Fig. 31.1). This reduction in cases together with the exclusion of high-risk bovine materials from the human food chain has resulted in a dramatic decrease in the number of human deaths in the United Kingdom, but inadvertent transmission of vCJD by transfusion of blood products has been reported. The extent and duration of the vCJD outbreak cannot be determined accurately, because of the lack of a suitable blood test for the disorder, and the prolonged incubation period of some prion protein genotypes in individuals, however a study of 32,441 archived appendix specimens in the United Kingdom showed that 16 of these samples harbored prion aggregates, indicating a remarkably high prevalence of 1 in 2000 individuals.
Properties of Prions
Classification
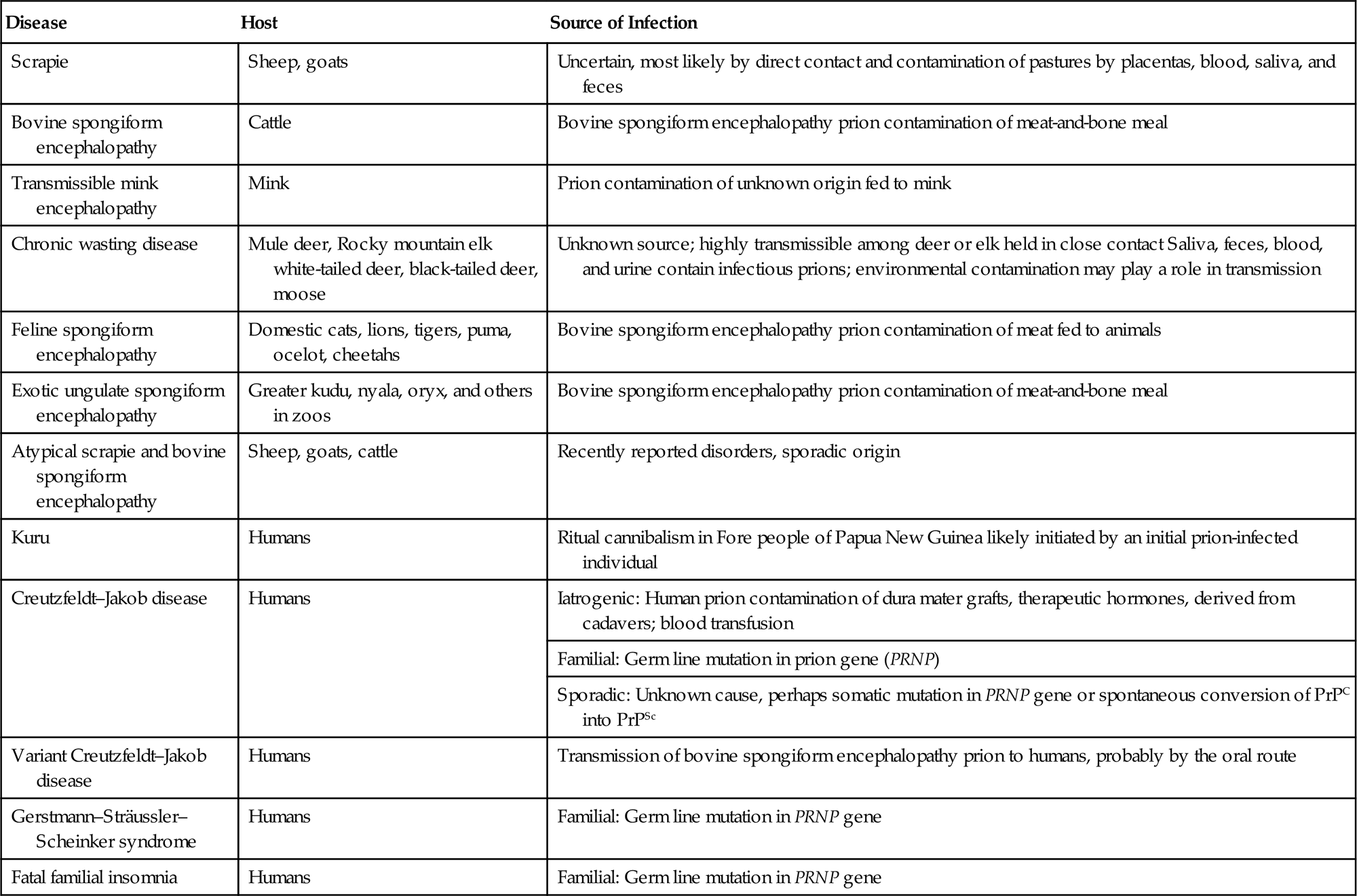
Prions have not been classified in the same way as viruses, thus there are no families, genera, or species. They first are identified by their host species, clinical disease, and their associated lesions (Table 31.1), and then characterized further by their molecular and biological properties. Their primary amino acid sequence mainly reflects the host from which they were isolated, but also registers mutations that define inherited variants—eg, in familial Creutzfeldt–Jakob disease in humans. Full amino acid sequences of virtually all important prion variants have been determined in different susceptible species and, as described below, naturally occurring amino acid substitutions are associated with relative susceptibility and incubation time in sheep and cervids (deer, elk, and moose).
Table 31.1
Prion Diseases of Animals and Humans
| Disease | Host | Source of Infection |
| Scrapie | Sheep, goats | Uncertain, most likely by direct contact and contamination of pastures by placentas, blood, saliva, and feces |
| Bovine spongiform encephalopathy | Cattle | Bovine spongiform encephalopathy prion contamination of meat-and-bone meal |
| Transmissible mink encephalopathy | Mink | Prion contamination of unknown origin fed to mink |
| Chronic wasting disease | Mule deer, Rocky mountain elk white-tailed deer, black-tailed deer, moose | Unknown source; highly transmissible among deer or elk held in close contact Saliva, feces, blood, and urine contain infectious prions; environmental contamination may play a role in transmission |
| Feline spongiform encephalopathy | Domestic cats, lions, tigers, puma, ocelot, cheetahs | Bovine spongiform encephalopathy prion contamination of meat fed to animals |
| Exotic ungulate spongiform encephalopathy | Greater kudu, nyala, oryx, and others in zoos | Bovine spongiform encephalopathy prion contamination of meat-and-bone meal |
| Atypical scrapie and bovine spongiform encephalopathy | Sheep, goats, cattle | Recently reported disorders, sporadic origin |
| Kuru | Humans | Ritual cannibalism in Fore people of Papua New Guinea likely initiated by an initial prion-infected individual |
| Creutzfeldt–Jakob disease | Humans | Iatrogenic: Human prion contamination of dura mater grafts, therapeutic hormones, derived from cadavers; blood transfusion |
| Familial: Germ line mutation in prion gene (PRNP) | ||
| Sporadic: Unknown cause, perhaps somatic mutation in PRNP gene or spontaneous conversion of PrPC into PrPSc | ||
| Variant Creutzfeldt–Jakob disease | Humans | Transmission of bovine spongiform encephalopathy prion to humans, probably by the oral route |
| Gerstmann–Sträussler–Scheinker syndrome | Humans | Familial: Germ line mutation in PRNP gene |
| Fatal familial insomnia | Humans | Familial: Germ line mutation in PRNP gene |
Certain biological properties are used to distinguish strains of prions, particularly scrapie strains. Following intracerebral injection of prion-containing material into several strains of inbred mice, the following parameters are recorded: (1) incubation period and mortality pattern; (2) distribution and extent of spongiform lesions, prion protein (PrP) plaques and astrogliosis in brains (assayed by immunohistochemical staining using anti-PrP and antiglial fibrillary acidic protein (GFAP) antibodies); (3) (in some cases) titer of infectivity in brains. Prion strains “breed true,” giving highly reproducible results in this kind of biological assay system. For example, prions from cattle, nyala, kudu, and domestic cats behave the same when subjected to this strain characterization protocol, indicating that all have been derived from the same source, namely cattle. Further, mice inoculated in the same way with material from cattle with bovine spongiform encephalopathy and humans with vCJD have behaved the same, yet differently from mice inoculated with material from sporadic cases of Creutzfeldt–Jakob disease.
Similar results have been recorded by biochemical analysis of prions recovered from various sources; for example, when human brain specimens were treated with proteinase K and their protease-resistant fragments were subjected to western blot analysis, seven different blot patterns were found. Six patterns represented sporadic Creutzfeldt–Jakob disease in humans; the seventh represented all cases of vCJD, which was similar to bovine spongiform encephalopathy in cattle.
Prions from animals and humans can also be transmitted to various other animals (hamsters, rats, ferrets, mink, sheep, goats, pigs, cattle, monkeys, and chimpanzees), although the “species barrier” typically results in prolonged and highly variable incubation periods. Additionally, the clinical signs, prion plaque morphology and distribution in the brain, and biochemical properties can change dramatically in a new species, indicating that a new conformational variant or “strain” has emerged. Subsequent passage of the new prion within the same host typically leads to a decrease in the incubation period, as prion conversion within the new host PrPC sequence becomes more efficient.
Prion Properties
Prions are normal cellular proteins that have undergone a pathologic conformational change that occurs posttranslationally. The normal protein, called PrPC (the term for the normal cellular isoform of the prion protein), is composed of about 209 amino acids (Mr 33,000–35,000). It is highly conserved and encoded in the genome of mammals. PrPC is ubiquitously expressed, and reaches particularly high levels in neurons and follicular dendritic cells. The function of PrPC is unclear; it binds both copper and iron, but “knockout” mice lacking the gene for the protein appear normal when young. In contrast, aged “PrP knockout mice” develop a demyelinating polyneuropathy, indicating that neuronal PrPC expression plays an important role in myelin maintenance. The amino acid sequence of PrPC and the abnormal isoform of the protein, called PrPSc (a term derived from the scrapie isoform of the prion protein, but in general use for all prion diseases) are identical in a given host. In a prion-infected individual, only the conformation of PrPSc has changed, from a structure made up predominantly of α-helices to one made up predominantly of β-sheets. An additional feature of PrPSc is that it consists of many PrP molecules stacked together as an aggregate and in some cases a long, highly ordered fibril. Although the structure of PrPC has been well-defined for many species, the structure of PrPSc has proven challenging to solve due to the aggregated nature of PrPSc. A monoclonal antibody has been developed that can discriminate between normal and disease-specific forms of PrP. It specifically precipitates bovine, murine, and human PrPSc, but not PrPC, confirming the presence of a conformational epitope common to prions from different species that is linked to disease but differs from the normal isoform of the protein.
When a given animal prion is passaged in mice or hamsters, the amino acid sequence of the recipient PrPSc is that of the PrPC of the recipient, not the donor. In a susceptible host species, there may be different PrPSc conformations that can develop despite individuals having the same amino acid sequence of PrPC. Each different conformation can in turn result in a different lesion pattern and different incubation and mortality patterns. This conformational variability is part of the basis for the differentiation of prion strains. For example, prions from vCJD in humans have characteristics distinct from those in other types of Creutzfeldt–Jakob disease although the primary amino acid sequence is identical. The biochemical and histopathologic features of vCJD are similar to prions isolated from cattle, mice, cats, and macaques infected during the bovine spongiform encephalopathy epizootic in the United Kingdom. Hamsters infected with transmissible mink encephalopathy develop two different prion strains, in spite of an identical amino acid sequence.
PrPSc protein is very resistant to many environmental insults, chemicals, and physical conditions that would destroy any virus or microorganism (Table 31.2). PrPSc is also at least partially resistant to endogenous proteases, which is the key to the exponential accumulation of PrPSc aggregates in the central nervous system.
Table 31.2
Effects of Physical and Chemical Treatments on Scrapie Prion Infectivitya
| Treatment | Reduction of Infectivity |
| 1 M NaOH | 106–8 |
| Phenol extraction | 106 |
| 0.5% sodium hypochlorite | 104 |
| Histopathologic processing | 102.6 |
| 3% formaldehyde | 102 |
| 1% β-propiolactone | 101 |
| Ether extraction | 102 |
| Autoclave 132°C for 90 min | 107.4 |
| Autoclave 132°C for 60 min | 106.5 |
| Autoclave 121°C for 90 min | 105.6 |
| Boiling 100°C for 60 min | 103.4 |
| Heating 80°C for 60 min | 101 |
aComposite of several studies, therefore no untreated control value given.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


