Orthomyxoviridae
Abstract
This chapter describes the properties of orthomyxoviruses and features of the diseases they cause in animals.
Keywords
Orthomyxoviridae; Influenzavirus; Influenza; Isavirus
The family Orthomyxoviridae includes viruses with genomes composed of several (six to eight) segments of single-stranded RNA. The most important members of the family are the influenza viruses, which are included in four genera (Influenzavirus A, B, C, and D). Influenza viruses that are pathogenic to wild and domestic animals and birds are included in the genus Influenzavirus A, whereas viruses in the two other genera (B and C) circulate continuously in humans. Influenza A viruses infrequently are transmitted from their animal hosts to humans, but human epidemics and pandemics caused by influenza A viruses typically have no animal involvement beyond the initial incursion. Human influenza viruses sporadically are transmitted to swine, leading to establishment of virus lineages adapted to this host species. Continuing surveillance, therefore, is essential to identify and detect influenza A virus variants that are capable of infecting humans while they are still confined either to their animal host or to a limited number of human contacts.
Recent developments have advanced significantly the understanding of the biology of influenza viruses. First, increased virologic surveillance and the rapid characterization of viruses by sequencing have confirmed extensive genomic rearrangements between different influenza viruses. Secondly, the emergence of the highly pathogenic Eurasian H5N1 virus in Southeast Asia in 1997 and the H1N1 virus pandemic of 2009 led to establishment of worldwide surveillance programs to identify potentially pandemic influenza viruses in wild and domestic animal species. Lastly, widespread use of sensitive and specific RT-PCR assays that detect all influenza viruses has facilitated detailed surveillance without the need for virus isolation. Once a sample is identified as influenza A-positive with this assay, the infecting virus rapidly can be further characterized by gene-specific RT-PCR assays to determine its hemagglutinin/neuraminidase subtype and cultured for subsequent antigenic analyses.
Although the original isolation of an influenza virus did not occur until 1930 (from swine), associated diseases previously had been recognized in both animals and humans. Indeed, human influenza was described by Hippocrates some 2400 years ago. Human pandemics have occurred throughout history, and the “Spanish flu” pandemic of 1918 was especially dramatic. The causative agent of highly pathogenic avian influenza (HPAI), “fowl plague,” was recognized in the late 19th century as a filterable agent (ie, virus), but was not identified as an influenza virus until 1955. High-pathogenicity avian influenza (HPAI) virus was first isolated from wild birds in 1961—specifically, common terns (Sterna hirundo) in South Africa—but until recently this highly lethal virus has since been rarely detected in wild birds. Low-pathogenicity avian influenza (LPAI) virus was first isolated from wild birds in 1972, and such low-virulence viruses are common in aquatic species of wild birds. Aquatic birds (orders Anseriformes and Charadriiformes), especially ducks, shorebirds, and gulls, are the essential reservoir hosts of low-pathogenicity influenza A viruses (Table 21.1). Influenza viruses replicate in the intestinal and upper respiratory epithelium of these birds without producing overt disease, and are excreted in high concentrations in feces and oral secretions. The viruses efficiently are transmitted by the fecal–oral route, and migrating aquatic birds carry viruses between their summer and winter habitats, which may span continents. Feeding stops along the flyways during the migrations provide further opportunity for spread of the viruses to resident contact wild and domestic bird populations, and facilitate the continuing process of evolution of these viruses.
Table 21.1
Hemagglutinin Subtype Distributiona Between Different Birds (Class: Aves) and Mammals (Class: Mammalia)
| HA Subtypec | Host of Origin | |||||
| Mammalia | Aves | |||||
| Humans | Swine | Equine | Anseriformes (eg, dabbling ducks) | Charadriiformes and Procellariiformes (eg, shorebirds, gulls, seabirds) | Galliformes (Domestic Poultry) | |
| H1 | ++ | ++ | + | + | ++e | |
| H2 | (++)b | ± | + | + | + | |
| H3 | ++ | ++ | ++ | ++ | ++ | ++e |
| H4 | ± | ++ | + | + | ||
| H5 | ± | ± | + | + | ++b | |
| H6 | ± | ++ | + | + | ||
| H7 | ± | ± | (++)b | + | + | ++b |
| H8 | ± | ± | ||||
| H9 | ± | ± | + | ++ | ++ | |
| H10 | ± | + | + | + | ||
| H11 | + | ++ | + | |||
| H12 | + | + | ± | |||
| H13 | + | ++ | + | |||
| H14d | ± | |||||
| H15d | ± | ± | ||||
| H16 | + | |||||
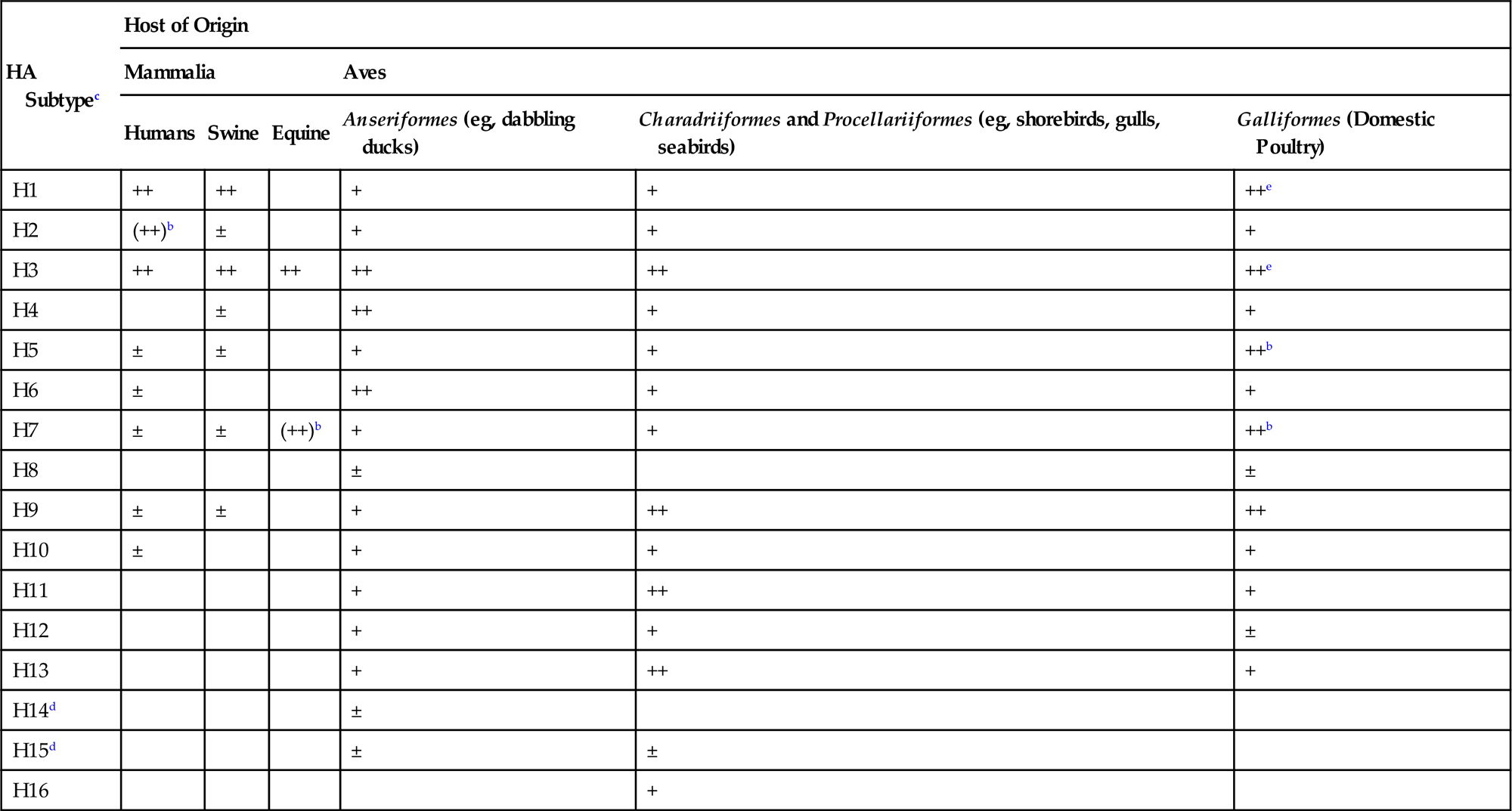
a±, sporadic; +, multiple reports; ++, most common.
b(), Previously common but now not reported.
ePrimarily swine influenza virus infections of domestic turkeys.
From Swayne, D.E. (Ed.). Animal Influenza. Copyright © John Wiley and Sons (2009), with permission.
Cross-species infections occur sporadically between birds and mammals, including swine, horses, dogs, mink, marine mammals, and humans (Fig. 21.1). Incursions of influenza A virus from wild aquatic birds into domestic poultry occur much more frequently, but until recently most of these events went undetected as the immediate consequences were limited. There have been only a very limited number of outbreaks of highly pathogenic avian influenza in domestic poultry worldwide that resulted in high death losses or regulatory action to eradicate or manage the infection, such as the recent epizootic of Eurasian H5N1 HPAI virus infection. In addition, many human infections with low-pathogenicity H7N9 avian influenza virus reported by China in 2013 complicate surveillance due to the lack of poultry mortality as a driver of diagnostic activities. Domestic swine are considered an important intermediate (“bridge”) host in those areas of the world where there is frequent contact between poultry and swine, although the premise that swine are an essential intermediate host for the development of pandemic influenza virus strains has not been substantiated. Whole-genome sequencing of influenza viruses is now widely used to reconstruct the phylogeny of each of the eight viral gene segments. For instance, comparison of the hemagglutinin (HA) and neuraminidase (NA) genes shows divergence into multiple subtypes (H1 through H16 and N1 through N9, respectively) and host-specific lineages within some subtypes. However, interspecies transmission and reassortment can complicate the interpretation of phylogenetic trees. In contrast, analyses of the matrix protein gene (M) of field strains of influenza A virus show two major avian lineages (North American and Eurasian), two equine lineages, two gull lineages (North American and European), two swine lineages (North American and Eurasian) and a human lineage. Analysis of the PB1 gene segregates human viruses between the North American swine and Eurasian avian groups. Predictably, there are exceptions such as an outbreak of equine influenza in China in 1989 that was caused by an H3N8 virus from a contemporary Eurasian avian source, whereas the classical H3N8 equine lineage has its origin from a North American avian lineage virus. Influenza A viruses from wild birds are very diverse, comprising the majority of the viral gene pool, and all gene segments evolve constantly. However, the virion surface genes accumulate amino acid changes most frequently. Evolution is detected not only in mammalian (eg, equine) and domestic poultry viruses, but also in viruses from wild birds.
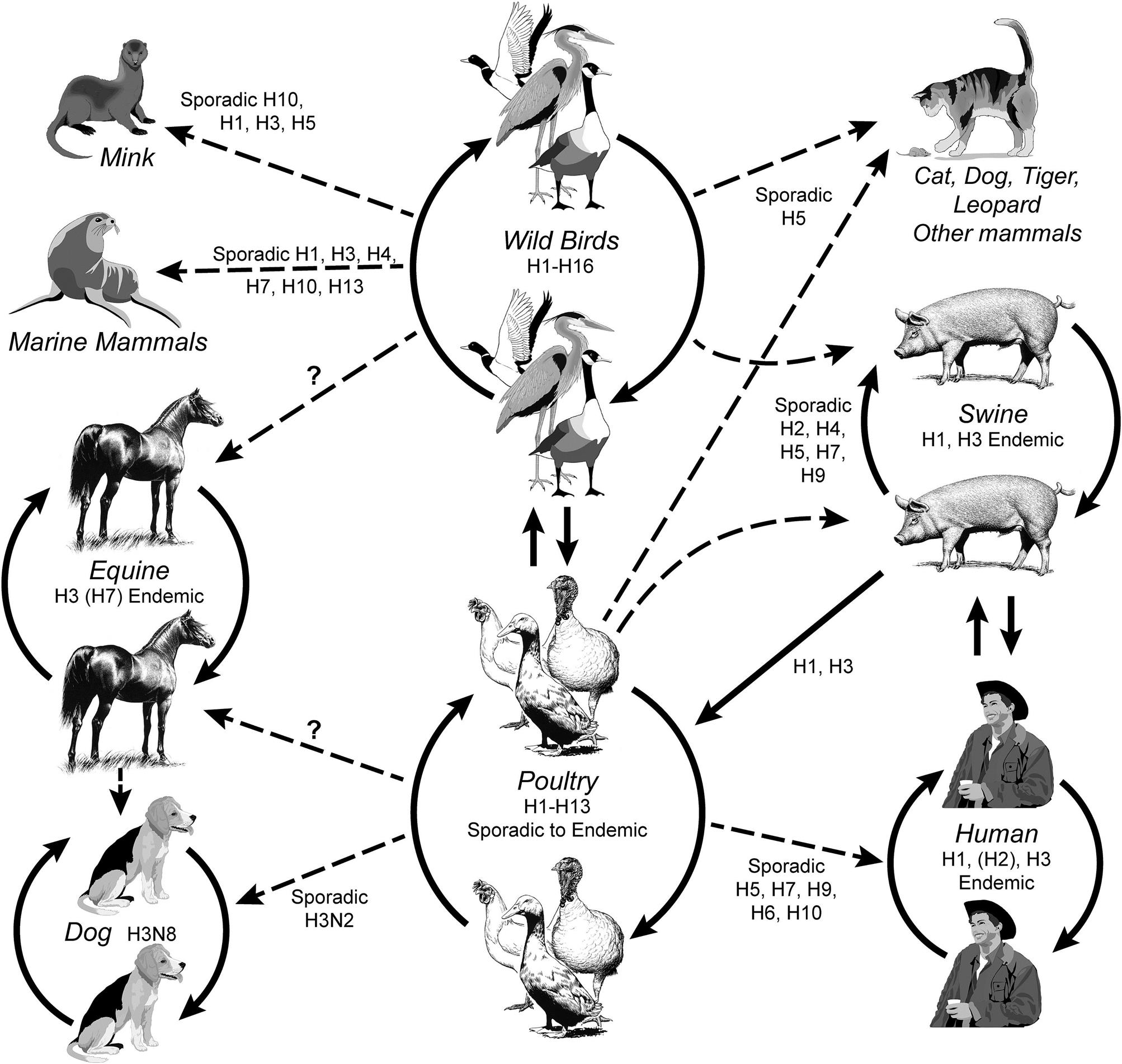
With renewed appreciation of the natural history of influenza viruses as “species jumpers,” prevention efforts logically will continue to focus on those situations in which high densities of birds and mammals are maintained in close proximity and in which there are rapid turnover in populations, such as live-animal markets. The latter often bring together a wide variety of poultry and other birds such as chickens, ducks, turkeys, pheasants, guinea fowl, and chukars as well as mammals, such as pigs, rabbits, and civets, without biosecurity practices. Live bird markets amplify, diversify (reassort), and perpetuate viruses within them, and serve as a source of infection of poultry farms via the movement of empty cages and personnel. Surveillance also is required for early identification of emerging reassortant viruses. Although improved agricultural biosecurity practices that separate swine and domestic poultry production from wild aquatic birds can reduce the frequency of newly emergent influenza virus variants, such approaches are often difficult to enforce, and the potential threat of epidemics of human influenza that emerge from avian or mammalian reservoirs will persist.
The need for intensive surveillance for the emergence of novel influenza viruses was demonstrated dramatically in April 2009, with the appearance of a new pandemic strain of influenza A virus in Mexico. Instead of the widely anticipated emergence of a new pandemic virus based on the highly pathogenic Eurasian H5N1 influenza virus, this newly emergent virus had its origin in triple reassortant swine viruses that had been circulating in pigs in North America since the late 1990s. The novel feature of this new virus was an additional reassortment that replaced two gene segments (NA and M) of the North American swine virus with the respective segments of the Eurasian swine virus. The origin and the date of development of this novel H1N1 virus have not been definitively established, as the first indication of its existence was from human infections. Initial investigations were unable to detect the novel H1N1 infection in swine in the absence of contact with infected workers. This recent event further emphasizes the unpredictable evolution and emergence of influenza virus as a mammalian pathogen.
Properties of ORTHOMYXOVIRUSES
Classification
The family Orthomyxoviridae comprises the genera Influenzavirus A, Influenzavirus B, Influenzavirus C, Thogotovirus, Quaranjavirus, and Isavirus. The name of the family is derived from the Greek myxa, meaning mucus, and orthos, meaning correct or right. The name was intended to distinguish the orthomyxoviruses from the paramyxoviruses. Influenza is the Italian form of Latin, from influentia, “influence,” so used because epidemics were believed to be caused by astrological or other occult influences. Influenza A viruses are common pathogens of horses, swine, humans, and domestic poultry throughout much of the world, but they also are the cause of sporadic or geographically limited infections and disease in mink, seals, whales, and dogs. Influenza B viruses are pathogens of humans, but there are reports of influenza B virus infection in seals. Influenza C viruses infect humans and swine, and reassortants have been detected, but influenza C viruses rarely cause serious disease in either species. The thogotoviruses are tick-borne viruses that sporadically infect livestock and humans in Africa, Europe, Asia, and most recently North America, but their pathogenic significance remains conjectural. The recently established genus Quaranjavirus includes viruses isolated from ticks and birds predominantly, but sometimes also from humans with febrile disease. The sole member of the genus Isavirus is infectious salmon anemia virus, a highly fatal disease of marine-farmed Atlantic salmon. Newly discovered orthomyxoviruses from cows (proposed new genus Influenzavirus D) and bats are awaiting definitive taxonomic classification.
A classification system was developed for influenza viruses because of the practical need to assess the risk represented by the emergence of new variant viruses, and the need to determine herd or population immunity against previously circulating strains so that vaccine requirements can be assessed. The emergence of variant viruses depends not only on genetic drift—ie, point mutations (nucleotide substitutions, insertions, deletions), but also on genetic shift—ie, genomic segment reassortment. Previously, drift and shift in only the viral hemagglutinin and the neuraminidase were intensively monitored, but, with the advent of enhanced sequencing technology, other viral genes may assume more importance in assessing risk. In the current classification system, influenza A viruses are categorized into 16 hemagglutinin (H) and 9 neuraminidase (N) subtypes, although recently described bat influenza viruses may increase the number of hemagglutinin sub types. In naming virus strains, influenza virus type (A, B, or C), host if other than humans (swine, equine, chicken, turkey, mallard, etc.), geographic origin (at province or state level), strain number, year of sample collection, and hemagglutinin and neuraminidase subtypes are included. Thus the full identification of an influenza virus looks like a secret code but is precise and informative. Examples of virus strain names include: A/equine/Miami/1/1963 (H3N8), the prototypic equine influenza virus 2; A/swine/Iowa/15/1930 (H1N1), the prototypic strain of swine influenza virus; A/Hong Kong/1/1968 (H3N2), the virus that caused the human pandemic of 1968; A/chicken/Scotland/1959 (H5N1), the first HPAI virus of the H5 subtype.
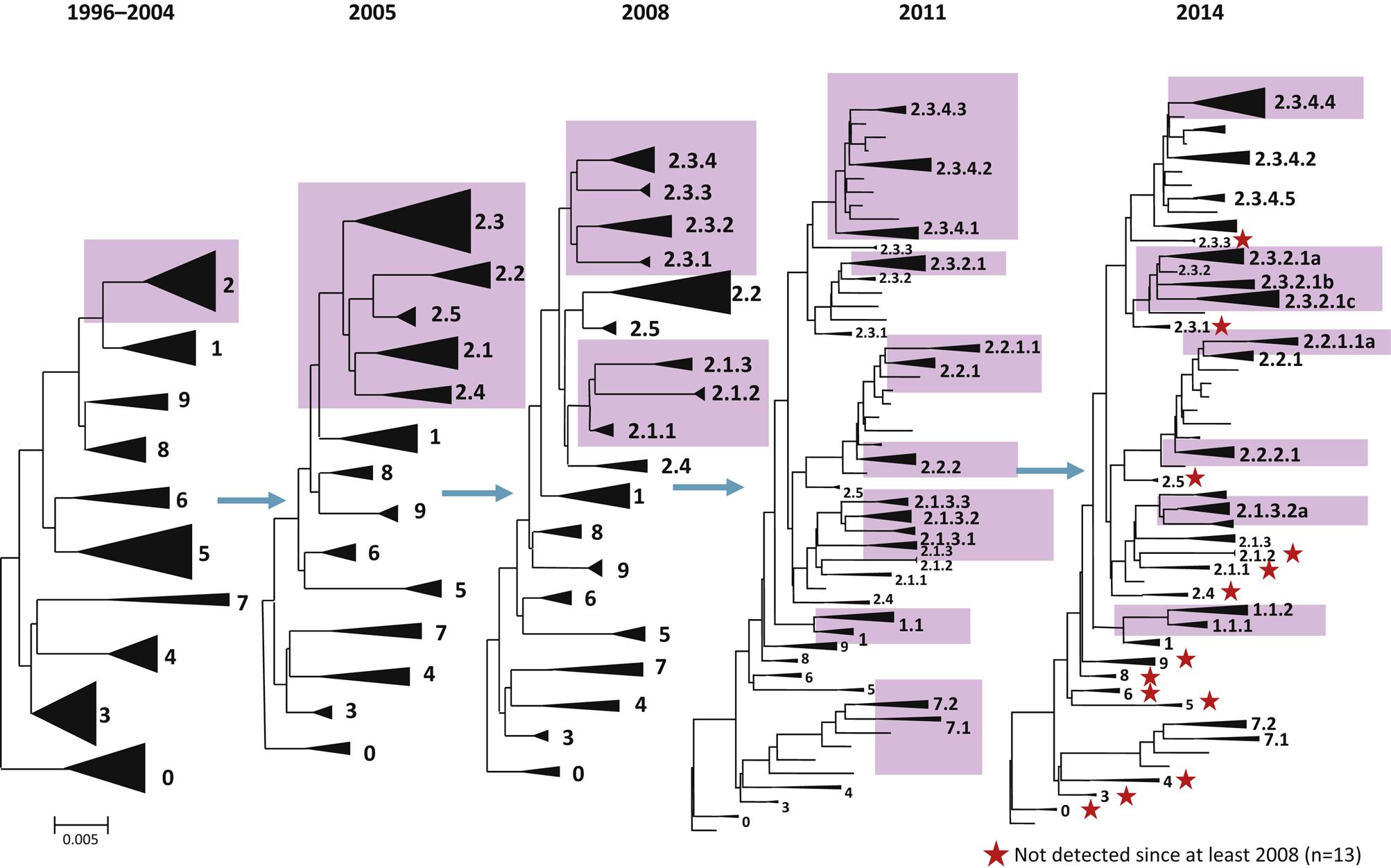
Changes recently have been implemented in the nomenclature for Eurasian H5N1 hemagglutinin, and for other influenza A viruses as appropriate, because the linking of the isolates to a specific geographic location becomes confusing and uninformative as the virus spreads globally. A numerical clade system has been adopted to better relate the evolutionary changes in these related H5N1 isolates over time; a clade is a taxonomic group comprising a single common ancestor and all descendants of that ancestor. For the Eurasian H5N1 hemagglutinin gene, the reference isolate is A/Goose/Guangdong/1/1996 (H5N1). The initial outbreak viruses from Hong Kong from 1997 were included in a single clade with the prototype virus, based on the hemagglutinin sequence. However, since 2003 the viruses have spread to progressively more regions beyond China and have evolved into several independent but related clades. By 2004, 10 distinct first order genetic clades were recognized (0–9), and continued evolution in subsequent years has resulted in a total of 30 additional second, third, and fourth order clades (eg, 2.1, 2.2, 2.1.3, 2.2.1, 2.1.3.2 and 2.2.1.1, etc.) (Fig. 21.2). This clade nomenclature system readily identifies the genetic linkage of the virus regardless of the geographic location, source of the isolate, or year of the isolate. The strain nomenclature system will continue to be maintained in repositories and be used to identify sequences deposited into databases such as GenBank.
Although any gene constellation and any combination of HA and NA genes can arise by genetic reassortment, only a limited range of combinations are recognized as important and naturally occurring subtypes responsible for animal infections: (1) enzootic H7N7 and H3N8 viruses (previously designated equine influenza viruses 1 and 2, respectively) that cause respiratory disease in horses; (2) enzootic H1N1, H1N2, and H3N2 viruses that cause influenza in swine; (3) sporadic H7N7 and H4N5 viruses that cause respiratory and systemic disease in seals; (4) sporadic H10N4 viruses that cause respiratory disease in mink; (5) historically endemic H1N1, H2N2, H3N2, and more recently sporadic or limited H5N1, H7N3, H7N7, H7N9, and H9N2 viruses that cause respiratory disease in humans; (6) geographically restricted H3N8 and H3N2 viruses that cause respiratory disease in dogs; (7) nearly all genetic combinations occur in wild aquatic birds, but particular emphasis is placed on detection of H5 and H7 viruses in domestic poultry because they can be associated with the high-pathogenicity phenotype (HPAI viruses).
Virion Properties
Orthomyxovirus virions are pleomorphic, filamentous but become spherical upon laboratory cultivation, and 80–120 nm in their smallest dimension (Fig. 21.3). They consist of a lipid envelope with large glycoprotein spikes surrounding eight (genera Influenzavirus A, Influenzavirus B, and Isavirus), seven (genus Influenzavirus C), or six (genera Thogotovirus and Quaranjavirus) helically symmetrical nucleocapsid segments of different sizes (Table 21.2). For influenza A and B viruses, there are two kinds of spikes: homotrimers of the hemagglutinin glycoprotein and homotetramers of the neuraminidase glycoprotein. Influenza C viruses lack a distinct neuraminidase spike, and have a single kind of glycoprotein spike that consists of multifunctional hemagglutinin-esterase molecules. Infectious salmon anemia virus (genus Isavirus) also has a hemagglutinin-esterase and a fusion or F protein. Regardless of the configurations, at least three functions are linked to the surface proteins: receptor binding, receptor cleavage, and membrane fusion. Virion envelopes are lined by the matrix protein, M1 on the inner surface of the lipid bilayer with a small number of interspersed ion channels composed of tetramers of the second matrix protein, M2. Genomic segments consist of a molecule of viral RNA enclosed within a capsid composed of helically arranged nucleoprotein. Three proteins that make up the viral RNA polymerase complex (PB1, PB2, and PA) are associated with the genomic RNA and nucleoprotein (NP). The genome consists of six to eight segments of linear negative-sense, single-stranded RNA, and is 10–14.6 kb in overall size. The genome segments have nontranslated regulatory sequences at both the 5′ and 3′ ends. The 13 nucleotides at the terminal 5′ end and 12 at the 3′ end are identical for each of the genomic segments and show partial inverted complementarity. This feature is essential for RNA synthesis.
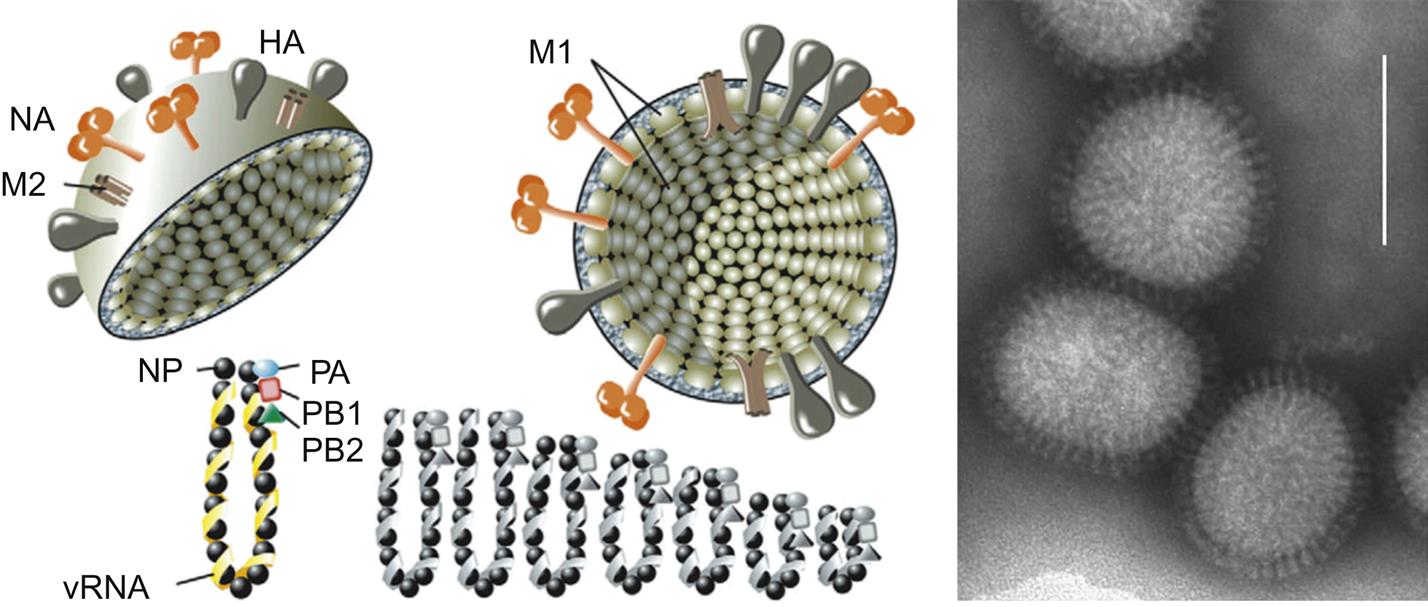
Table 21.2
Properties of Influenza Viruses
Six genera: Influenzavirus A, Influenzavirus B, Influenzavirus C, Thogotovirus, Isavirus, and Quaranjavirus
Virions are pleomorphic, spherical, or filamentous, 80–120 nm in diameter, and consist of an envelope with large spikes surrounding six to eight helically symmetrical nucleocapsid segments of different sizes
The genome consists of linear negative-sense, single-stranded RNA, divided into six to eight segments, 10–14.6 kb in overall size
There are two kinds of spikes (influenza A & B virus); rod shaped, consisting of homotrimers of the hemagglutinin glycoprotein, and mushroom shaped, consisting of homotetramers of the neuraminidase protein
Transcription and RNA replication occur in the nucleus; capped 5′ termini of cellular precursor mRNAs are cleaved and used as primers for mRNA transcription; budding takes place on the plasma membrane
Defective interfering particles and genetic reassortment occur frequently
Because of their structural and biochemical properties, influenza viruses are sensitive to heat (56°C, 30 minutes), acid (pH 3), and lipid solvents, and are thus very labile under ordinary environmental conditions. However, infectious influenza A virus has been recovered after 30 days in cold lake water.
Virus Replication
Influenza virions attach to cells via the binding of their activated hemagglutinin to sialic-acid-containing receptors on the plasma membrane, as depicted and described for influenza A virus (Fig. 21.4). Different host cells have surface glycans with different linkages of N-acetyl neuraminic acid (sialic acid) to a galactose residue, and the hemagglutinin recognizes these different linkages, which in turn determine the host range of the virus. The gut epithelium of ducks has a receptor with an α2,3 linkage (SAα2,3Gal), whereas the predominant influenza virus receptor in the upper respiratory tract of humans is an α2,6 linkage (SAα2,6Gal). There is also evidence that binding affinity for the SAα2,6Gal glycan varies with the length of the oligosaccharide. Human-adapted H1 and H3 viruses show binding preference for long oligosaccharides present on epithelial cells in the upper respiratory tract, and mutations that affect this binding alter transmissibility. A single amino acid change in the hemagglutinin protein of the 1918 H1 Spanish flu virus at position 190 (E190D) changes binding preference from SAα2,3Gal to SAα2,6Gal.
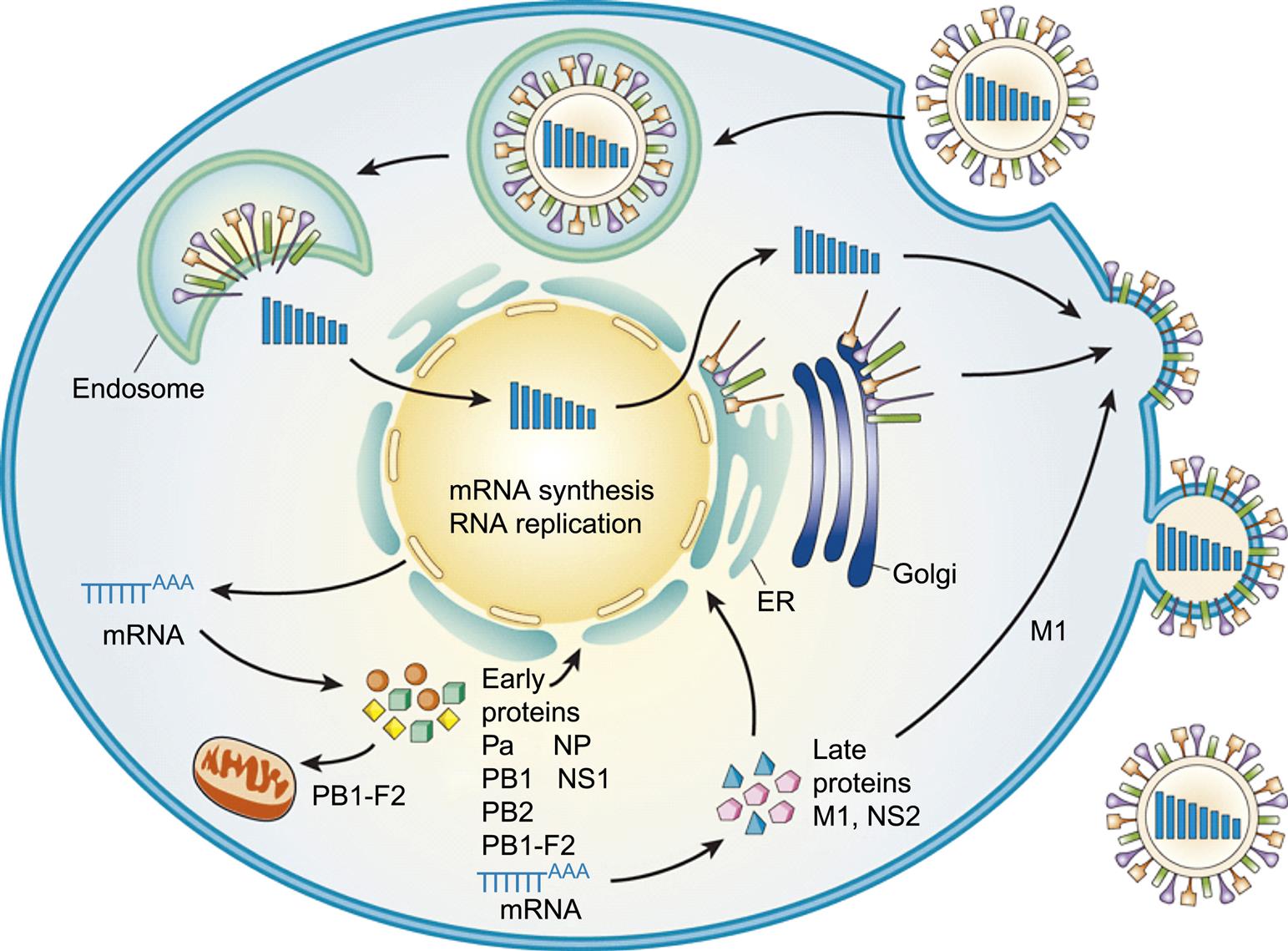
Influenza viruses enter cells via receptor-mediated endocytosis. While in the endosome, the ion channel M2 (matrix protein 2) tetramer allows the flow of protons into the virus particle to enable dissociation of the other matrix protein (M1) from the RNP complex (which includes viral genomic RNA, nucleoprotein, and polymerase proteins), thus freeing it from the viral envelope. The low pH (acidic) of the endosome triggers a conformational change in the hemagglutinin (HA) protein such that the hydrophobic domain of the HA2 trimer mediates fusion of the viral envelope with the endosomal membrane, releasing the RNA, nucleoprotein, and polymerase proteins (RNP) into the cytoplasm (Fig. 21.4). Amantadine and rimantadine inhibit virus infection by blocking the ion channel activity of M2. A unique feature of influenza virus is that all RNA synthesis takes places in the nucleus of the cell. This requires that the RNP, because of its size, be actively transported into the nucleus. Nuclear localization signals on the nucleoprotein interact with the nuclear transport machinery of the cells to transport the RNP into the nucleus.
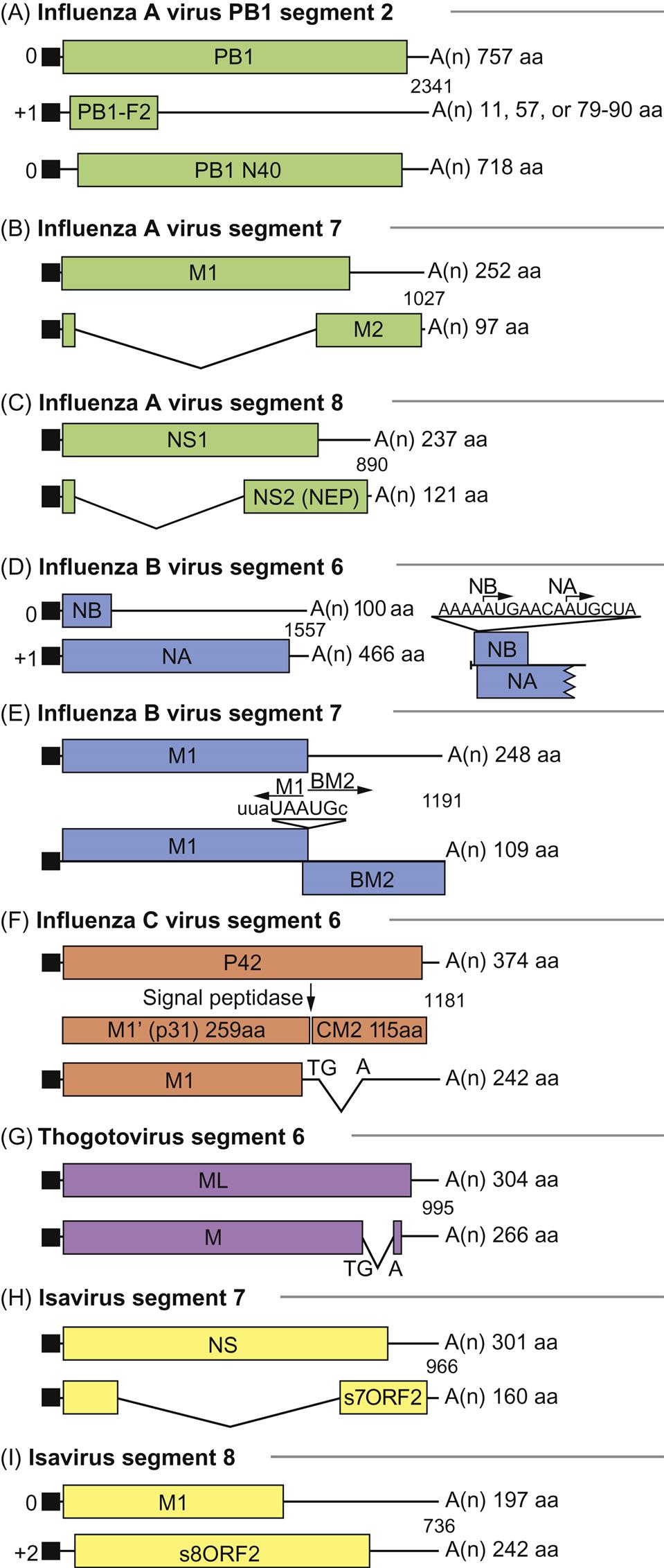
As with all viruses with negative-sense RNA genomes, the genome of orthomyxoviruses serves two functions: as a template for the synthesis of messenger RNAs (mRNAs), and as a template for the synthesis of positive-sense replicative intermediate RNA, which is the template for progeny genomic RNA synthesis. Primary transcription involves an unusual phenomenon known as cap snatching: the viral endonuclease activity of the polymerase (PA) cleaves the 5′-methylguanosine cap plus about 10–13 nucleotides from cellular precursor mRNAs that are captured by PB2, another component of the polymerase complex. These caps are then used by the virus as primers for transcription by the viral RNA polymerase (transcriptase; PB1). The viral mRNAs thus are capped and also become polyadenylated through template slippage and repeated transcription of five to seven “U” residues on the virion RNA. All orthomyxoviruses extend the coding capacity of their genomes by producing two proteins from certain genes by using a splicing mechanism. Influenza A virus uses splicing for gene segments 7 (M1 and M2) and 8 (NS1 and NEP/NS2) (Fig. 21.5); influenza B virus uses gene segment 8 (NS1 and NEP/NS2); influenza C virus uses gene segments 6 (CM1 and CM2) and 7 (NS1 and NEP/NS2); thogotoviruses use gene segment 6 (M and ML); isavirus uses gene segments 7 and 8. The NS1 and NS2 (HEP) proteins are defined as nonstructural proteins whereas the various M, CM, and ML proteins are all membrane associated. Influenza B virus also uses a different strategy for gene segment 7, involving overlapping stop and start codons to generate two protein products. In certain influenza A virus strains, a PB1-F2 protein of 87–90 amino acids is generated by a ribosomal +1 reading frameshift.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


