1 Principles of Drug Therapy
The intent of drug therapy is to induce a desired pharmacologic response for a sufficiently long period of time while preventing adverse drug events (see Chapter 4). For most drugs the magnitude of pharmacologic response is proportionately related to the (log of) drug concentration at the tissue (receptor) site (Figure 1-1). Understanding the relationship among dose, drug concentration, and response requires an understanding of the pharmacodynamics (i.e., the science of drug action), or the physiologic and biochemical effects of a drug and their relationship to the drug’s mechanism of actions. Most commonly, the response is measured in the animal but may also occur in a microbe or parasite.1 Pharmacodynamics may be studied in vitro (isolated cells or tissue), ex vivo (isolated cells or tissues after exposure to the drug in the intact animal), or in vivo (exposure and study occurs in the whole animal). Pharmacodynamics ultimately should be integrated with the science of pharmacokinetics, that is drug movements through the body.
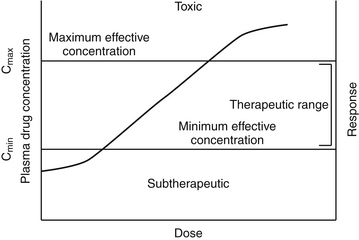
Dose–Response Relationship
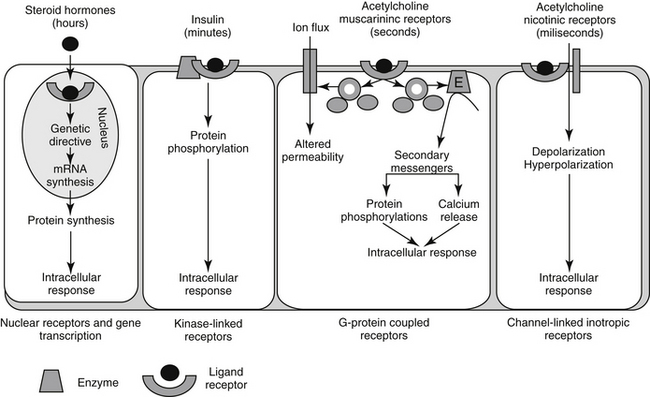
Pharmacodynamic responses occur at many levels, ranging from single molecules to whole animals. Drugs induce their responses through a number of mechanisms, most of which involve direct or indirect interactions with cell macromolecules and generally proteins. Direct interactions with nonprotein molecules are less common, but examples include nucleic acids (e.g., cancer chemotherapeutic agents), metal chelating drugs, or antacids used to chemically neutralize gastric acid. Drug responses more commonly reflect the interaction of the drug, acting as a ligand, with receptors (Figure 1-2). A receptor most commonly is a large protein macromolecule (e.g., structural, enzymatic, carrier, or ion channel proteins) responsible for cellular signaling. Receptors may be located on or in the cell membrane, in the cytosol, or within an intracellular structure (i.e., nucleus). Physiologic functions of the body generally are regulated by multiple receptor-mediated mechanisms, each responding to different molecular stimuli. Examples of target receptor categories include hormones, neuromodulatory receptors, and neurotransmitters.
Interaction between a drug and its receptor generally results in activation (either directly or indirectly) of cellular biochemical processes (e.g., ion conductance, protein phosphorylation, or DNA transcription) by way of a transduction pathway that ultimately brings about the pharmacologic effect.1 A time lag may be associated with transduction (Figure 1-2). Activation reflects the drug’s mechanism of action, whereas the sequelae of the stimulation at the molecular, cellular, or tissue level reflect the drug’s pharmacodynamic effects. Secondary intracellular messenger molecules are often activated by drug–receptor interaction; they subsequently set in motion a cascade of events that eventually causes the response. Among the most common receptors are transmembrane receptors linked to guanosine triphosphate–binding proteins (G proteins). These then activate second messenger systems such as adenylyl cyclase (e.g., beta-adrenoceptors), the cytosolic inositol triphosphate pathway (e.g., alpha-adrenoceptors), or membrane-bound diacylglycerol (DAG).
Two properties describe drug-receptor interactions: affinity, or the capacity of a drug to bind to a receptor, and intrinsic efficacy (or activity), or the capacity of a drug to activate or inactivate a receptor. The latter is a complex relationship dependent on drug concentration, receptor activation, and cellular response. Affinity is usually mathematically defined as the reciprocal of the dissociation constant of the drug for the receptor.1 A receptor often is characterized by multiple types (e.g., alpha- and beta-adrenergics; mu, kappa, and delta opioid receptors) and subtypes (e.g., alpha 1 and 2, beta 1, 2, or 3; mu 1 or 2). The selectivity of a drug action generally reflects the specificity of the drug for the target receptor binding. Drugs often target multiple receptors, although interactions may be concentration dependent or may result in blocking rather than activating specific receptors. Receptor characteristics (e.g., numbers and affinities) are influenced by both external and intracellular conditions. Tissues vary in receptor numbers and subtypes; indeed, proper physiologic responses are dependent on this variability. The relationship between a drug and pharmacologic response was at one time assumed to be directly proportional to the number of receptors occupied by the drug, with a maximal response reflecting 100% occupancy and activation (i.e., the drug receptor theory). However, the interactions are much more complex than that described by a simple linear relationship. Different kinetic relationships (linear, log-linear, polynomial), multiple activation states, (active, inactive, resting), cell amplification of the drug-receptor signal, and existence of spare receptors are some examples of the complexities that determine the relationship between drug concentration and response. The scientific description, examination, and ultimate prediction of drug–receptor interactions are often accomplished by generation of dose–response curves; the graphic representation most generally is represented by a sigmoidal curve (Figure 1-3; see also Fig. 1-1). The curve indicates that increasing drug concentration at the receptor site increases the number of bound receptors and thus drug effect. If concentration–response curves are plotted on a logarithmic (x) axis, the portion of the curve that lies between 20% and 80% of the maximal response is generally linear. This is the portion most relevant to therapeutic concentrations and thus encompasses the therapeutic range. Increasing a drug dose within this range generally results in a proportional increase in response. However, once the 80% mark is passed, as receptors become saturated, a much larger increase in concentration (dose) is necessary to increase response. Although this may increase the risk of adverse effects, for drugs which are safe, it may also prolong the dosing interval since the response will not change much as concentrations decline to the 80% level. At that point, drug concentration declines exponentially (generally first order), and response will also decline log-linearly with time (e.g., 50% decline with one half-life). Note that for some drugs (i.e., drugs that irreversibly interact with receptors, drugs with active metabolites,), response may not be related to plasma (tissue) drug concentrations.
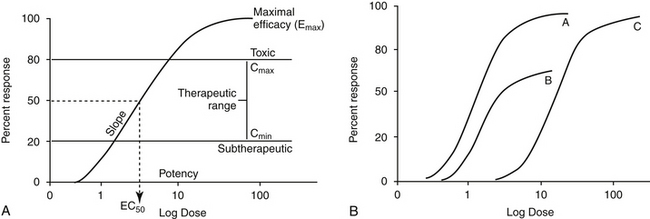
The interaction of a drug with a receptor is similar to that between a substrate binding to the active site of an enzyme. As such, similar equations and parameters are used to describe the relationship between dose and response. The effective dose (ED) is the quantity of administered drug that will produce the (desired) effects for which it is administered. The median effective dose (ED50) is the dose that produces the desired effect in 50% of a population. However, dose is a less accurate descriptor of what is happening at the receptor than is effective concentration (EC). The affinity of the drug for the receptor is described by the (effective concentration) EC50, the drug concentration that yields 50% of the maximal response (see Figure 1-3, A). The different actions of a drug, such as therapeutic and adverse effects, are often due to the drug binding to different receptors with different EC50 values. Ideally, the EC50 for an adverse event is higher than that of a therapeutic response. The ratio of adverse event EC50 to the therapeutic effect EC50 is the therapeutic index and provides some indication of drug safety in that the larger the index, the safer the drug. Efficacy and potency are two terms used to describe the relationship among drug, concentration, and response. Efficacy refers to the maximum effect (Emax) a drug can have (e = 1 indicates a full response), whereas potency is a comparative term that describes the concentration of two drugs necessary to induce the same magnitude of response. Generally, plots describing efficacy and potency are based on log 10 concentration versus percent response curve. (see Figure1-3, A). Drugs are considered equal in efficacy if they can cause the same magnitude of response; the more potent drug will cause an EC50 at a lower concentration. As such, the dose of a less potent drug may simply need to be increased to achieve the same effect of the more potent drug.
Drug–receptor interactions do not always yield the maximal response. Receptor interaction with a drug that acts as an agonist (generally, a structural analog of the targeted receptor) results in some level of activation. The occupation theory of drug–receptor interaction indicates that the magnitude of the response produced by an agonist is directly proportional to the number of receptors occupied.1 Pharmacodynamic antagonism occurs when one drug inhibits the agonistic effects of another through actions at the same pathway, although this does not have to occur at the receptor. Like an agonist, an antagonist interacts selectively with receptors, but it lacks intrinsic efficacy and thus is able to block or reduce the action of an agonist at the receptor. Drugs that target opioid receptors (at least three types, with mu receptors having at least two subtypes) offer an example of the different sequelae of drug–receptor interactions. The “full” agonist (e.g., fentanyl) results in the maximal effect (e.g., at mu receptors). A drug that acts as a “reversal” agent would reduce the effect of an exogenous agonist (e.g., naloxone reverses the effects of fentanyl) whereas a “blocking” drug antagonizes endogenous agonists (e.g., catecholamines targeted by beta- or alpha-blockers). Drugs might have dual agonist and antagonist properties at the same receptor (although to variable degrees in different tissues). Because the response is not maximal, they are referred to as partial (low-efficacy) agonists; buprenorphine is a partial agonist opioid at mu receptors. Drugs might also act as an agonist at some receptors and an antagonist at other receptors. Butorphanol is a mixed agonist/antagonist (at kappa and mu receptors, respectively).
The relationship between an antagonist and receptor can be described as competitive or noncompetitive. Competition between compounds occurs at the same receptors and can be reversible or irreversible. Reversible antagonists easily dissociate from the receptor, whereas irreversible antagonists form a stable chemical bond with the receptor (e.g., in alkylation). If the interaction is reversible, an agonist present at sufficiently high concentrations can displace an antagonist. As such, reversal of the agonist or the response may require a higher dose (or a repeat dose) of the antagonist (see Figure 1-3, B). In contrast, an irreversible antagonist cannot be displaced from the receptor. As such, the cellular response can not occur until the receptor is replaced (duration dependent on rate of receptor turnover) and any remaining unbound antagonist has been removed from the body. The presence of a reversible competitive inhibitor will decrease the potency of a drug because a higher concentration will be necessary to induce the same pharmacologic response.
In contrast, the presence of an irreversible competitive inhibitor will decrease the efficacy of the drug by preventing the maximal possible response. In contrast to competitive antagonists, noncompetitive antagonists interact with receptors at a site different from the agonist–receptor interaction site. The interaction often involves a site in the transduction pathway between the receptor and the pharmacodynamic response.1 Noncompetitive interactions are generally, but not always, reversible. Note that three other types of antagonism, in addition to pharmacodynamic, can occur between drugs: chemical antagonism results from a direct chemical interaction between two drugs (i.e., a weak acid and weak base), physiologic antagonism occurs when two drugs act in the same physiologic system but act on different receptors or pathways, and pharmacokinetic antagonism occurs when one drug alters the response to another drug through changes in disposition (see Chapter 2).
The pharmacodynamic response to a drug ideally will occur with any given dose within a therapeutic range. The therapeutic range provides a target for the dosing regimen. It consists of a minimum effective plasma drug concentration (PDC) (trough or Cmin), below which therapeutic failure is likely to occur, and a maximum effective PDC (peak or Cmax), above which a type A adverse reaction (see Chapter 4) is more likely to occur (see Figures 1-1 and 1-3).2,3 Dosing regimens are composed of a dose (e.g., mg/kg) and an interval (e.g., every 8 hours) for each route. The dose of the regimen generally is designed to achieve and maintain PDC within the therapeutic range throughout most of the dosing interval. Thus targeted peak PDCs often approximate but do not exceed Cmax, whereas trough concentrations approximate but generally do not drop below Cmin. However, a therapeutic range is a population statistic that describes the concentrations between which most animals will exhibit the (desired) pharmacodynamic response; each animal will respond (therapeutically or adversely) at a different point in the range. Although most animals will respond at some point within the range (the majority in the middle of the range), a small percentage will respond above or below the range. Therapeutic drug monitoring (see Chapter 5) is used to establish where in the therapeutic range the individual animal will respond; in other words, it will establish the patient’s therapeutic range. Ideally, studies that determine the dosing regimen in a target species reflect integration of pharmacokinetic and pharmacodynamic studies in that species.4 Two primary components of a dosing regimen are dose interval. In general, dose, which ultimately determines PDC, is influenced primarily by the tissue that dilutes the drug (i.e., volume of distribution), whereas interval is influenced by elimination of the drug (i.e., half-life). Unfortunately, studies that describe the time course of a drug in animals are limited and, when available, generally focus on healthy rather than diseased animals. As such, clinicians are faced with individualizing dosing regimens in the patient according to the principles of clinical pharmacology—that is, the study of drugs5 and their behavior (disposition)6 in animals.
Determinants of Drug Disposition
Mechanisms of Drug Movement
Drugs move through the body by two major mechanisms: bulk flow and passive diffusion. The cardiovascular system is the primary determinant of bulk flow; glomerular filtration is one of the more important specific examples. The chemical nature of the drug does not affect bulk flow. Most drugs are characterized by a molecular weight (MW) of 350 or less, ensuring movement of unbound drug between endothelial cells despite the presence of protein filters in the endothelial gaps. Exceptions are made for those tissues whose capillaries are not fenestrated, because endothelial cell junctions are tight. Drug movement for these tissues must occur across cell membranes. Drugs can move through cells directly through the lipid layers of the cell (e.g., passive diffusion), through aqueous pores formed by aquaporins (proteins) that span the width of the membrane, by combination with transmembrane carrier proteins, and by pinocytosis. Of these, transmembrane (passive) diffusion and carrier-mediated movement are most important.
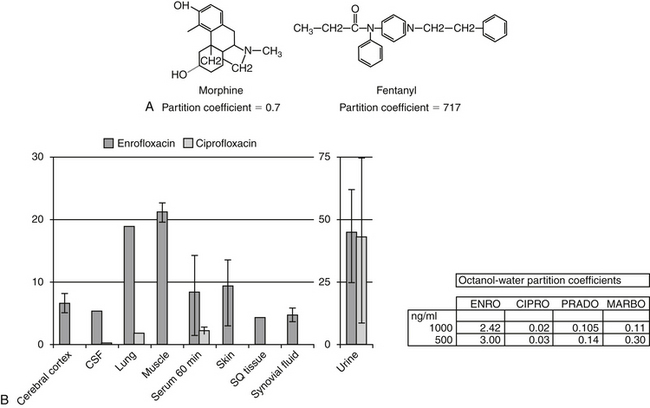
Passive diffusion occurs independent of any other mechanism of drug movement and requires no energy. Passive diffusion through cell membranes depends on a number of factors. The single most important factor is concentration of diffusible drug, which is most easily manipulated by increasing the dose. Additionally, host and drug factors will also influence the concentration of diffusible drug. Host determinants of passive diffusion are subject to change and include thickness of the membrane to be traversed (inversely proportional; e.g., edematous compared with normal tissues), surface area (directly proportional; e.g., small intestine versus stomach), environmental pH (see ionization), and temperature (directly proportional). In contrast, drug characteristics influencing passive diffusion are not subject to change, and thus cannot be easily manipulated. They largely influence lipid solubility of the drug.2,3 Lipid solubility is, in turn, influenced by a number of drug characteristics, including the inherent chemical structure of a drug, such as molecular weight (smaller molecules diffuse more easily) and partition coefficient (PC). The PC is an experimental measure of relative lipid solubility as influenced by the chemical structure of the drug. The ratio is determined by mixing the drug in a combination of water and an organic solvent (e.g., octanyl). The difference in drug concentration between the two solvents once mixing is complete reflects, in part, the inherent lipid versus water solubility of the drug. Measurement of the concentration of drug in each solvent generates an octanyl:water coefficient or PC, which might be useful for predicting the ability of a drug to pass through cell membranes. A ratio greater than 1 suggests greater distribution to the organic phase, indicating lipid solubility. For example, in regards to providing analgesia, fentanyl (PC of 717) is both more potent and more rapid acting, but shorter in duration, compared to morphine (PC of 0.7), presumably because it can move more rapidly through the blood–brain barrier (Figure 1-4). Benzene rings, carbon double bonds and methyl groups tend to make a drug more lipid soluble, whereas polar compounds such as amine or hydroxyl groups contribute to drug-water solubility.
In addition to lipid solubility, drug pKa and environmental (host) pH will also influence distribution of drug into body compartments. The Henderson-Hasselbalch equation describes the pH partition theory (Figure 1-5). Assuming a drug is sufficiently lipid soluble to cross cell membranes, a steady-state equilibrium will be reached when the amount of drug moving from one area equals the amount moving in the opposite direction and the concentration of diffusible drug will be equivalent on either side of a membrane. However, in its ionized form, a drug is not diffusible, cannot traverse lipid membranes, and becomes trapped and thus “partitioned” by the pH in its surrounding environment. Although the concentration of the nonionized (diffusible) portion on each side of the membrane will be equal (assuming a steady state equilibrium is reached), the total concentration on either side may differ if the pH on either side of the membrane is different (Figure 1-5). The difference depends on the pKa of the drug and the environmental pH on either side of the membrane. Drugs will be trapped by ionization when present in an “unlike” environment. Drug pKa (the pH at which the drug is 50% ionized and 50% nonionized; ratio 1:1), and its behavior as a weak acid or base defines the degree of ionization. For a weak acid, as local pH decreases (becoming more “like”), the nonionized and thus diffusible proportion will increase. In contrast, for a weak acid, an increase in pH (“unlike”) will increase the ionized proportion. The opposite is true for a weak base: increasing pH will increase the nonionized or diffusible proportion, whereas a pH decrease will increase the ionized, nondiffusible portion (Box 1-1).
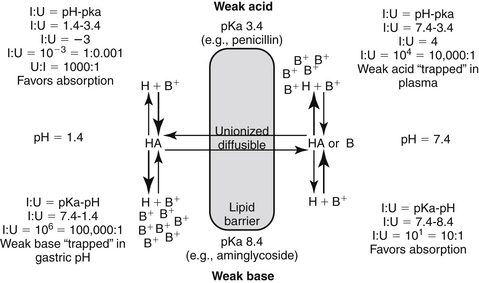
Figure 1-5 The Henderson-Hasselbalch equation and the pH partition hypothesis predict the behavior of an ionizable compound on the basis of its pKa and the ambient pH. A weak base (bottom) will be predominantly ionized in an acidic environment (i.e., gastric pH), and thus is likely to be trapped. It is less likely to be ionized in a higher pH and thus is more diffusible (i.e., plasma). In contrast, a weak acid (top) is likely to be nonionized in the acidic environment and thus is more likely than the weak base to move across the lipid membrane and be absorbed. However, in the plasma the higher pH will increase the proportion of ionized drug, limiting its diffusion from plasma into cells. (see Box 1-1.)
Box 1-1 The pH Partition Hypothesis
The pH partition hypothesis is the ratio of ionized:nonionized drug and is based on pH (of the environment) and pKa (of the drug) (see Figure 1-5). The proportion of ionized to unionized drug is described by the Henderson-Hasselbalch equation. For acids, the ratio (ionized:nonionized) is 10n, and for a weak base the ratio is 10-n. The ratio of the ionized:nonionized (I:U) drug can be useful for predicting movement between tissues. A drug is considered significantly ionized if the ratio (I:U) is greater than 100. Thus for a weak acid the drug is considered ionized in an environmental pH that is 2 or more higher than its pKa (pH − pKa = 2; 10n = 102 = 100 I : 1 U) A weak base is considered ionized if the pH is or 2 or more below its pKa.. (pH − pKa = -2; 10-n = 10-(-2) = 102 = 100). Ideally, for drug movement to occur by passive diffusion, the pH will be no more than 2 pH units above the pKa for the acid and no more than 2 pH units below the pKa for the base. The gastrointestinal tract (pH 6.0) offers an example of how pH partition influences drug movement. Orally administered aminoglycosides (weak bases with pKa approximating 7 to 9) are ionized at a ratio of approximately 1000:1 (6 − 9 = −3, 10 -n = 10 –(-3), = 103 = 1000:1 [I:U]. Thus there is only one nonionized or diffusible ion for each 1000 ionized amikacin molecules. The ionized molecules will be trapped and will not be absorbed, limiting absorption of this water-soluble drug. In contrast, the proportion of the ionized weak acid penicillin (pKa about 2) would be 0.0001:1 (2 − 6=10-4); for every ionized molecule, 10,000 molecules would be nonionized (the inverse of 0.0001:1). As such, if other factors are supportive (e.g., the drug is not destroyed by the gastric acidity), penicillin should be well absorbed from the gastrointestinal tract. The urine is another site where pH partition may influence drug movement. Penicillin located in urine with a pH of 7 would be ionized at a ratio of 10,000:1 (105), whereas the aminoglycoside would be ionized at a ratio of only 100:1. If the urine pH was 8, the ratio of I:U would be 10 for the aminoglycoside, which is not considered significant to preclude drug movement. In either pH the aminoglycoside would be more likely than the penicillin to be passively resorbed and to penetrate microbial membranes. Drug that is partitioned by ionization acts as a reservoir, replacing nonionized drug that may leave the other side of the membrane in an open system. Eventually, assuming the nonionized side remains “open,” both sides of the membrane will eventually be depleted of drug.
Although less common, drug movements other than passive diffusion and bulk flow influence PDC. Carrier-mediated transport includes both facilitated diffusion (nonactive) or active transport. An example important transport system is the P-glycoprotein system, an MDR-1 gene product best known for imparting multidrug resistance to cancer cells (and to microbes).7 However, this transport system occurs in several tissues in the body, including renal tubular brush borders and bile canniculi (responsible for drug excretion from the body); the brain or other “sanctuaries” (responsible for keeping exogenous componds out of critical tissues); characterized by a blood–tissue barrier; and in portals of entry, including the lower gastrointestinal tract (reducing oral drug bioavailability).8 Pinocytosis is a rare drug movement exemplified by the uptake of vitamin B12 in the ileum and aminoglycosides by renal tubular cells.
Plasma Drug Concentrations
Although response to a drug reflects concentrations at the tissue (or cellular level), because tissue samples cannot be collected easily, drug concentrations at the tissue site are approximated by measuring PDCs. After administration of a fixed dose of a drug, several drug movements act in concert to determine PDC (Figure 1-6).2,3,6,9 These movements largely, but not exclusively, depend on passive diffusion of the drug and include absorption (A) from the site of administration to systemic circulation, defined as the major vessels and well-perfused organs; distribution (D) of the drug from systemic circulation to tissues (target and nontarget) and back again; and elimination of the drug from the body by metabolism (M) and excretion (E). These drug movements are dynamic, occurring simultaneously, and their net effects determine PDC at any time during the dosing interval after administration of a fixed dose. Because passive diffusion is the major determinant of each drug movement, each in turn is influenced by a number of other factors. The impact of these factors on the drug movements and the time course of drug in the body as a result of these movements can be described or modeled mathematically, leading to the study of pharmacokinetics. Each drug movement is influenced by host and drug factors that may affect therapeutic success (see Chapter 2).
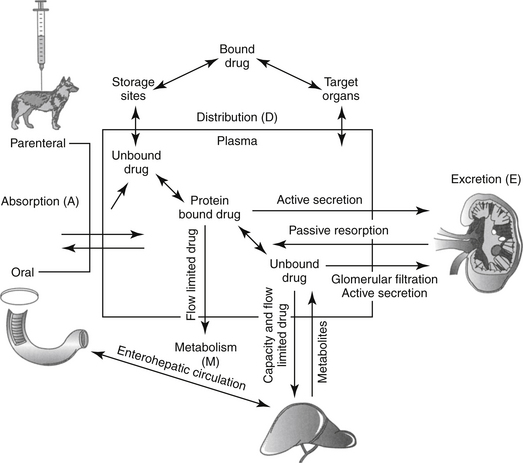
Absorption
Bioavailability, extent, and rate of absorption
The percentage of an administered dose of drug that reaches systemic circulation and is thus able to induce a response is referred to as bioavailability (F).2,3,9,10 Bioavailability (see Pharmacokinetics section) is used to predict drug efficacy after different routes of administration or administration of different formulations of the same drug. Absolute bioavailability or extent of absorption is the actual bioavailability and can be determined only by comparing the appearance of the drug preparation to that after intravenous administration of the drug. In contrast, relative bioavailability of two different preparations or by two routes of administration of the same drug can be evaluated by comparing their area under the curve (AUC).10 The area under the concentration versus time curve (AUC) describes the entire time course of the drug. Because its magnitude depends on maximum drug concentration and rate of elimination, it is influenced by several drug movements, including absorption. The AUC is used to calculate several other pharmacokinetic parameters; clinically, it is useful for determining response to certain drugs, particularly antimicrobials.11,12
Oral absorption
Most orally administered drugs reach systemic circulation after absorption from the small intestine. The rate and extent of drug absorption in the gastrointestinal tract depend on a number of host factors, most of which affect passive diffusion (Figure 1-7).9 These include gastrointestinal pH, which favors absorption of weak acids; surface area, which favors absorption in the small intestine compared with the stomach; motility, which mixes the drug, the concentration of diffusible drug at the site of movement; permeability and thickness of the mucosal epithelium; and intestinal blood flow, which maintains the concentration gradient across the mucosal epithelium. The latter factor of blood flow is important only for drugs capable of rapid transfer across the epithelium.
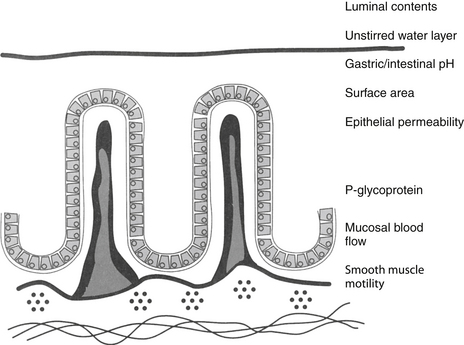
In general, the intestinal surface area is so large that changes seldom are of sufficient magnitude to effect absorption (see Chapter 2). However, changing particle size of the drug preparation may cause differences in the rate or extent of absorption and markedly different bioequivalences. Drug preparations often are specifically designed to alter rates of absorption by manipulation of particle size.
Bioavailability of an orally administered drug also is influenced by factors after it passes into the gastrointestinal epithelium. Bioavailability is decreased if the drug is metabolized by intestinal epithelial cells, microbes, or by the liver. Additionally, enterocytes can decrease oral bioavailability of a drug by causing its efflux from enterocytes, in part because of the presence of active transport proteins located on enterocyte cell membranes.13
Transport membranes may act alone or in concert with one another; for some drugs, one transporter may predominate, but for others, none may. Identifying the role of each transporter protein is complicated and difficult to study. Several families have been identified. (1) Transporters located on the apical cell membrane include the family of organic anion-transporting polypeptides (OATPs); these proteins are also located in the liver, kidney, and brain and influence distribution and excretion. These proteins transport a large number of amphipathic drugs (e.g., digoxin, steroids) and endogenous compounds (e.g., steroids, thyroid hormones). (2) Proton-dependent oligopeptide transporters (POTs), driven by a proton gradient, are present in the kidney, brain, and the gastrointestinal tract, where their activity appears to increase from the duodenum to the ileum. Among the drugs influenced by these transporters are beta-lactam antimicrobials, ACE-inhibitors, and prodrugs of selected antivirals. (3) The family of ATP-binding cassette (ABC) superfamily of transporters includes the P-glycoproteins. These include multidrug resistance protein (MRP1) and a multispecific organic anion transporter (MOAT) also located in other tissues. An efflux transporter, P-glycoprotein is characterized by broad substrate specificity. Because it is abundant in enterocytes, it can profoundly decrease the oral bioavailability of target drugs. It is associated and shares substrate specificity with selected cytochrome-P450 enzymes (most notably, CYP3A), which decreases drug that is not effluxed from the cell by the transporter (see Chapter 2).13
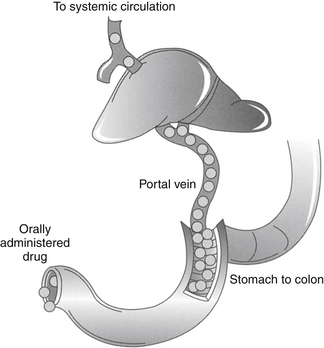
Hepatic metabolism also can profoundly affect the PDC of an orally administered drug. After gastrointestinal absorption, drugs enter the portal vein and then the liver (Figure 1-8). As such, an orally administered drug is exposed to hepatocytes before it enters the systemic circulation. Drugs characterized by a high hepatic extraction ratio (>70% extracted) are almost completely removed from the blood by hepatocytes during the first passage of blood through the liver. As a result, after oral administration, drugs that undergo first-pass metabolism may not reach systemic circulation in concentrations sufficiently high to cause a pharmacologic response. Despite good to excellent oral absorption, such drugs are characterized by poor bioavailability and are administered either parenterally (e.g., lidocaine) or in oral doses high enough to compensate for first-pass metabolism by the liver. Examples of drugs that are orally administered yet undergo significant hepatic first-pass metabolism include selected cardiac drugs (e.g., propranolol and other beta-blockers, diltiazem in some species, hydralazine, nitroglycerin), diazepam, and opioid analgesics. The negative effects of first-pass metabolism on pharmacologic response may be reduced if the drug metabolites (e.g., propranolol and diazepam) are also pharmacologically active.
Distribution
Once a drug reaches the systemic circulation, it must be distributed from the central (blood) compartment to peripheral tissues to impart a pharmacologic effect. Further, drug in tissues will be distributed back into plasma so that it can be eliminated. The major factors that determine drug distribution to and from tissues include drug lipid solubility and its ability to penetrate cell membranes, the degree to which the drug is bound to plasma or tissue proteins, and regional (organ) blood flow. The presence of transport proteins (p-glycoprotein, OATPs, and POTs) also influence drug distribution, particularly in “sanctuary” tissues characterized by tissue–blood barriers (e.g., brain, cerebrospinal fluid, placenta).13
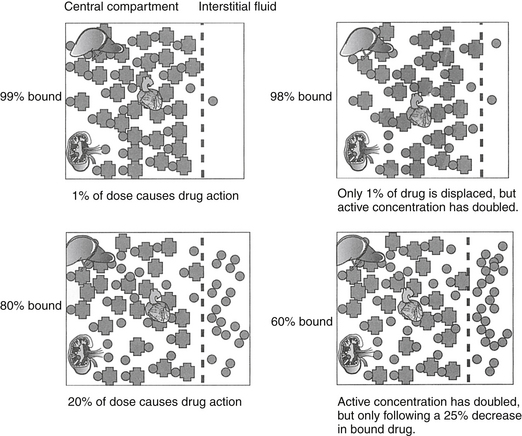
Binding of lipid-soluble drugs to plasma proteins facilitates their circulation. Drug binding to proteins may influence several determinants of drug movement, including distribution.14 Weakly acidic drugs tend to bind to albumin, whereas weakly basic drugs tend to bind to α1‑glycoproteins.15 The large molecular weight of protein precludes movement of bound drug from circulation into tissues. As such, the protein-bound drug is not pharmacologically active; cannot be filtered in the normal glomerulus, and for some drugs (i.e., capacity-limited, discussed later), cannot reach drug metabolizing enzymes in the hepatocyte (or other organ). Presumably, drugs that are highly protein bound may be more likely to be involved in adverse reactions early in the dosing regimen because displacement of only a small proportion of drug from the protein (e.g., due to competition with other protein-bound drugs or hypoalbuminemia) can increase the total amount of free, active drug (Figure 1-9).2,3 A drug is considered significantly (highly) protein bound if 80% or more bound; for a drug bound less than 80%, displacing enough drug to significantly increase the unbound proportion is difficult. For example, displacement of only 1% of a drug that is 99% protein bound (e.g., nonsteroidal antiinflammatories) can double the concentration of pharmacologically active drug. In contrast, to double the pharmacologically active form of a drug that is only 80% bound (i.e., going from 20% to 40% unbound) would require displacement of 25% of the bound drug, which is clinically difficult to achieve. Yet even with highly protein-bound drugs, the clinical relevance of displacement and increased concentrations of unbound drug is questionable: in the face of normal renal and hepatic function, clearance of the freed drug by these organs will increase such that “extra” drug is rapidly removed.16 However, it is not clear that clearance will increase sufficiently in the face of organ dysfunction (e.g., renal or liver disease). Further, the package insert for Converia, which is highly protein bound, indicates that the plasma drug concentration of several other highly protein bound drugs increased with co-administration.
In addition to its ability to reach target tissues and organs of elimination, distribution of a drug is important because of its influence on PDC and drug elimination. The amount of tissue to which a drug is distributed, often estimated by the theoretical parameter, volume of distribution (Vd) of the drug, directly but inversely influences PDC.2,3,17 Because it is theoretical, Vd is generally referred to as apparent Vd. The Vd of a drug also describes the ease with which the drug leaves the plasma. Simplistically, volume of distribution is also the volume to which a drug would have to be distributed if it were present throughout the body in the same concentration as that measured in the plasma after intravenous (IV) administration of a known dose. It can be exemplified by adding 5 g of dextrose (considered the dose) to each of two beakers containing a different but unknown volume of water. The dextrose is allowed to distribute equally throughout the beaker (no membranes are present in the beaker) and then the concentration is measured. If the concentration in beaker A is 5% (50 mg/mL or 5 g/100 mL), then the volume of water in the beaker must be 100 mL. If the concentration in beaker B is 2.5% (25 mg/mL or 25 g/100 mL), the volume must be 200 mL. The Vd in beaker A is twice that in beaker B. The Vd of a drug in an animal is determined essentially the same way: a known dose is given intravenously to ensure that all the drug reaches the site being measured, the drug is allowed to distribute until equilibrium (or pseudo-equilibrium) is reached, and the peak drug concentration is determined. The Vd is then calculated by dividing the dose by the peak concentration (the y intercept, Co, A or B): Vd = Dose/Co (see the Pharmacokinetics section).
The PDC (and response to) a drug at a known dose varies inversely with Vd. Vd differences among species and ages (pediatric versus geriatric) can dramatically affect PDC (see Chapter 2). In addition, diseases associated with fluid retention or obesity are likely to increase the Vd of many drugs (and thus decrease PDC), whereas dehydration or weight loss is likely to decrease Vd and thus increase PDC (see Chapter 2). The impact depends upon whether the drug is distributed to total body water (i.e., a lipid-soluble drug) or extracellular fluid (i.e., a water-soluble drug). Because the distribution of a drug to peripheral tissues removes drugs from organs of drug clearance, Vd also directly and proportionately influences elimination half-life (Figure 1-10). Thus any factor that increases Vd will tend to decrease PDC but prolong the elimination half-life and thus the presence of the drug in the body.
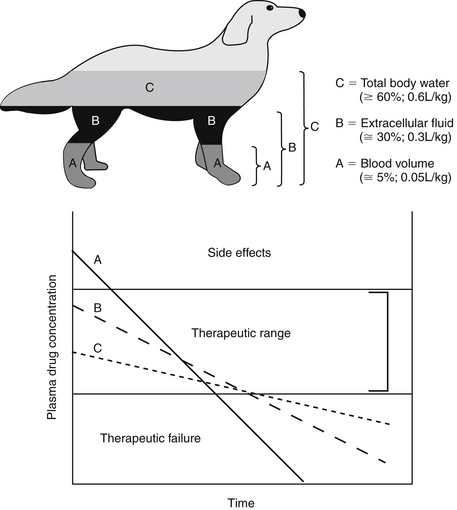
Capillaries of selected organs, including the brain, cerebrospinal fluid, eye, testis, and prostate, are not fenestrated, and drugs must diffuse through the capillary endothelium to penetrate these tissues.18,19 For treatment of such organs, lipid-soluble drugs (i.e., Vd >0.6 L/kg) are more likely than water-soluble drugs to penetrate the endothelial barrier and achieve therapeutic concentrations. However, simply because a drug has a Vd that equals or exceeds the volume of TBW does not assure lipid solubility nor does it guarantee sufficient tissue distribution. Drug distribution to various tissues differs with drug chemistry, and intracellular accumulation may occur for many reasons. Ion trapping was previously discussed; drugs also can bind to intracellular proteins (i.e., Na+ K+ ATPase of digoxin), bone (e.g., Ca2+ or Mg+ by tetracyclines) or other macromolecules (e.g., lysozymes by aminoglycosides). Bound drug is generally not active (unless binding occurs to target ligands), but bound drug may act as a reservoir for interstitial and plasma drug concentration (e.g., the antimicrobial cefovecin). Ion trapping or binding of a drug will remove it from plasma; as such, as PDC decreases, for any given dose, the Vd will increase. The Vd of a drug characterized by such multicompartments may result in a Vd that is greater than the volume of the animal (e.g., >1 L/kg). Such a drug exemplifies the theoretical nature of Vd in that it simply (mathematically) describes the amount of tissue that is diluting the drug if the concentrations measured in plasma were the same throughout the body. Although Vd is a useful parameter, particularly for calculating drug dose, it should not be used to indicate where (i.e., ECF, ICF, TBW, or selected tissues) a drug has distributed. Further, because the rate and extent of drug distribution to tissues varies, the time to equilibrium also will vary. Calculations for Vd (and determining peak PDC) generally should not occur until distribution equilibrium has been reached.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


