CHAPTER 24 Primary Hyperaldosteronism
Primary hyperaldosteronism (PHA) has been recognized as the leading cause of endocrine-mediated hypertension in human beings. Since Conn’s first association of mineralocorticoid excess with an adrenocortical tumor,1 hyperaldosteronism also has been attributed to primary idiopathic adrenal hyperplasia, defects in synthesis and metabolism of adrenal steroids, and rare instances of renin-secreting tumors. In the past decade, a series of case reports in cats has raised awareness of PHA as a cause of hypertension and hypokalemia, with the possibility of added complications of excesses of other adrenal steroids. The knowledge base from human medicine will be an invaluable asset as veterinarians improve the ability to diagnose and manage this disorder.
THE RENIN-ANGIOTENSIN-ALDOSTERONE SYSTEM AND CLASSIFICATION OF HYPERALDOSTERONISM
The renin-angiotensin-aldosterone system (RAAS) acts to maintain the volume of extracellular fluid, circulatory pressure, and electrolyte homeostasis through integrated effects of enzymes and hormones, chiefly on the vasculature and kidney (Figure 24-1), and continues to be a subject of review in the scientific literature.2–4 The juxtaglomerular cells that line afferent arterioles of the renal glomeruli both synthesize prorenin and remove a segment of its N-terminus to form active renin, which is stored in secretory granules. Both prorenin and renin are released from juxtaglomerular cells, the former in a tonic pattern and the latter in response to regulated pathways.5 Active release of renin is stimulated predominantly by decreased renal perfusion detected by baroreceptors in the afferent arterioles. Other stimuli for renin release include a decrease in sodium content in glomerular filtrate, as detected by the macula densa cells of the proximal tubule, and sympathetic nerve stimulation via β-adrenergic receptors. In the circulation, renin acts on angiotensinogen, a protein synthesized in the liver, to cleave a decapeptide, angiotensin I, from the N-terminus. A peptidase, angiotensin-converting enzyme, removes two amino acids from the C terminal of angiotensin I to form angiotensin II, which has potent biological effects. The principal effects of angiotensin II arise from its binding to type 1 receptors to mediate vasoconstriction, promote renal tubular reabsorption of sodium, stimulate release of aldosterone from the adrenal cortex, and exert negative feedback on the release of renin.

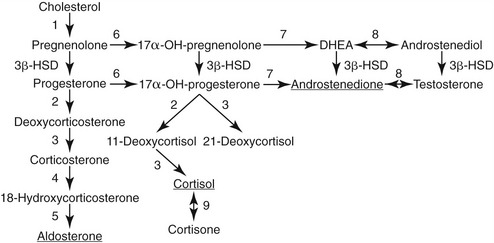
Aldosterone is synthesized from cholesterol through a series of intermediary metabolites including progesterone, 11-deoxycorticosterone, and corticosterone (Figure 24-2) in the zona glomerulosa of the adrenal cortex. In addition to stimulus from angiotensin II, aldosterone is released in a direct response to hyperkalemia and to a lesser effect of adrenocorticotropic hormone (ACTH) from the anterior pituitary gland. After secretion into the circulation, aldosterone enters target tissues and exerts its effects through binding to mineralocorticoid receptors, which have affinity for, and are activated by, both cortisol and aldosterone. Mineralocorticoid-responsive tissues of epithelial origin (e.g., renal tubule, colon, sweat glands, and salivary glands) express the enzyme 11β-hydroxysteroid dehydrogenase type 2, which converts cortisol to cortisone, a steroid that does not bind to the mineralocorticoid receptor and enables access by aldosterone to the receptor. In the kidneys aldosterone acts on the distal collecting tubule and collecting ducts to stimulate activity of epithelial sodium channels, promoting sodium reabsorption with concurrent loss of potassium and hydrogen ions. Therefore, when stimulated, the classic cascade of events in the RAAS results in an increase of circulatory pressure via vasoconstriction and retention of sodium. Mineralocorticoid receptors in nonepithelial tissues have minimal 11β-hydroxysteroid dehydrogenase 2 activity and may be maintained in a tonic inactivated mode when occupied by cortisol.6 The role of these receptors and aldosterone-mediated effects in inflammation and fibrosis occurring with cardiac, renal, and vascular disease, and metabolic syndrome is under active investigation.6–9
Hyperaldosteronism, or increased aldosterone secretion, can arise from a primary or secondary origin. PHA refers to excessive production from autonomous adrenal dysfunction. The ensuing effects of sodium retention and hypertension result in suppression of renin and angiotensin. Secondary hyperaldosteronism refers to increased aldosterone production in response to increased stimulation by the renin-angiotensin system. In most instances secondary hyperaldosteronism arises as a compensatory physiological response to counteract dehydration, hypotension, or sodium deficiency (reduced intake or increased loss). However, pathological manifestations of mineralocorticoid excess can arise from conditions of secondary hyperaldosteronism, notably from reduced renal perfusion secondary to renal disease or from rare instances of renin-secreting tumors.10
OVERVIEW OF PRIMARY HYPERALDOSTERONISM IN HUMAN BEINGS
ADRENAL ADENOMA AND IDIOPATHIC HYPERPLASIA
Three extensive reviews of these disorders have been published recently and serve as the main source of information for this discussion.11–13 Adrenal adenoma and idiopathic hyperplasia account for more than 95 per cent of the diagnoses of human hyperaldosteronism, with idiopathic hyperplasia being more common than adrenal adenoma (60 per cent versus 35 per cent). Both conditions can occur with unilateral (most adenomas) or bilateral (most hyperplasia) distribution. Risk factors to initiate screening for hyperaldosteronism include presence of refractory hypertension, unexplained hypokalemia, an adrenal mass, or a family history. Categorization of the lesion is based on histopathological examination of the adrenal cortex (hyperplasia versus microadenoma or macroadenoma). At the time of diagnosis more than 60 per cent of affected persons are normokalemic, with hypokalemia more likely to occur with adrenal adenomas.13–15
The aldosterone to renin ratio (ARR) is regarded as the most reliable screening test. Pretesting preparation includes an attempt to correct hypokalemia, an increase in sodium intake if on a restricted diet, and/or withdrawal of therapeutic agents that alter the ARR (e.g., mineralocorticoid receptor antagonists, potassium-sparing or -wasting diuretics, products containing licorice root) for at least 4 weeks.13 False-positive ARR results most commonly are a result of very low renin activity, which may be attributable to performance capabilities of the renin activity assay, potassium or sodium loading, administration of other medications, or presence of concurrent diseases. Tests to confirm PHA assess the response to treatments designed to suppress circulating aldosterone concentrations. At present the tests considered most commonly are oral sodium loading, saline infusion, fludrocortisone administration with sodium supplementation, and the captopril challenge test,13 but no confirmatory test is regarded as the “gold standard.” An apparent increasing prevalence of PHA has raised controversy, with some authors suggesting an epidemic while others caution against overestimation of its occurrence.12,16–20
When screening and confirmatory tests support the existence of PHA, diagnostic measures are employed for localization. Often the initial test is a computed tomography (CT) scan of the adrenal glands to identify or exclude a large mass characteristic of an adrenocortical carcinoma. Limitations with CT include inconsistency in distinguishing small adenomas (<1 cm) from bilateral idiopathic hyperplasia or detection of an incidental nonfunctional neoplasm when there is bilateral hyperplasia. If the patient is a candidate for surgery, and CT results are not clearly indicative of an adrenal mass, adrenal venous sampling (AVS) is the procedure of choice. A catheter is inserted in a femoral vein and threaded into the caudal vena cava. Samples are collected from the inferior phrenic vein adjacent to the opening of the left adrenal vein and from the right adrenal vein. A “peripheral” sample is collected from the iliac vein. Aldosterone and cortisol concentrations are measured in all samples. Comparison of cortisol and aldosterone results in the three samples provides, first, an indication of the success of adrenal vein catheterization and, second, information on whether there is asymmetry of aldosterone concentrations in adrenal venous samples. If a patient is not a candidate for AVS or does not have access to a center performing the procedure, adrenal scintigraphy provides a means for localization of aldosterone production. Current protocols use 131I-6β-iodomethyl-19-norcholesterol (NP-59) as the labeled agent. Patients are pretreated with dexamethasone and saturated potassium iodine to minimize uptake of NP-59 by adrenal cells for cortisol synthesis and to block thyroidal uptake of free 131I. A summation of the diagnostic performance of scintigraphy across 11 studies showed an overall 82 per cent diagnostic sensitivity, 78 per cent specificity, and 82 per cent accuracy in identification of aldosterone-producing adenomas.21 A limitation of this procedure is low uptake of labeled agent by small adrenal adenomas.
ADRENOCORTICAL CARCINOMA
Adrenocortical carcinomas account for less than 1 per cent of cases of PHA in human patients.11 The diagnosis is based on screening and confirmatory tests as outlined, and on identification of an adrenal mass (usually >4 cm) on CT examination. Adrenal carcinomas also may exhibit variation in steroidogenic production, with clinical signs related to mineralocorticoid excess only, or mixed effects of glucocorticoid, mineralocorticoid, and/or sex steroid production.22 Some carcinomas possess little or no 11β-hydroxylase activity, thus having minimal, if any, aldosterone secretion (see Figure 24-2). Signs of mineralocorticoid excess are caused by increased deoxycorticosterone production, leaving low or nondetectable circulating aldosterone concentrations, as has been reported in human beings and a dog.22,23
FAMILIAL CAUSES OF HYPERALDOSTERONISM
Familial hyperaldosteronism type I (glucocorticoid-remediable hyperaldosteronism) occurs in 0.5 to 1 per cent of cases of PHA. Affected persons usually are not hypokalemic but have a variable severity of hypertension. Suspicion for this disorder is raised by the presence of hypertension and cerebrovascular complications at a young age and a familial history of these occurrences. Initial confirmation of hyperaldosteronism is based on screening and confirmatory tests as described above. Findings of imaging studies and AVS testing are not unlike those of bilateral adrenal hyperplasia.24 The etiology is a hybrid gene derived from the 5′ regulatory sequence of the 11β-hydroxylase gene (CYP11B1) with the 3′ coding sequence from the aldosterone synthetase gene (CYP11B2).25–27 Therefore cells in the zona glomerulosa secrete aldosterone in response to ACTH rather than angiotensin II, and cells in the zona fasciculata produce hybrid steroids such as 18-hydroxycortisol or 18-oxo-cortisol. Measurement of 18-hydroxysteroids is the most definitive biochemical indicator in diagnosis.24 Familial hyperaldosteronism type I has an autosomal dominant mode of inheritance and a genetic test is available.26 Administration of dexamethasone results in decreases in glucocorticoid and mineralocorticoid concentrations, and blood pressure, serving as the basis for treatment of glucocorticoid-remediable hyperaldosteronism. Familial hyperaldosteronism type II (non–glucocorticoid-remediable hyperaldosteronism) refers to the familial occurrence of hyperaldosteronism owing to adrenocortical adenoma or hyperplasia. Genetic studies of families in Australia, Italy, and South America have identified a linkage of these causes of hyperaldosteronism with chromosome 7p22.28
Apparent mineralocorticoid excess is a rare disorder in which hypertension and hypokalemia are identified in young children. Plasma renin activity and circulating aldosterone concentrations are low. The etiology is a result of inactivating mutations of the gene encoding 11β-hydroxysteroid dehydrogenase type 2, which are inherited as an autosomal recessive trait. The mutations allow cortisol to gain access to the mineralocorticoid receptor.29 The biochemical marker for this disorder is reduction of serum concentrations of cortisone and its metabolites in relation to cortisol. Nephrocalcinosis has been reported in association with apparent mineralocorticoid excess.29,30 Restriction of dietary sodium is of benefit in controlling hypertension. Other therapeutic measures include administration of combinations of potassium supplementation, mineralocorticoid receptor antagonists, or hydrochlorothiazide.
FELINE PRIMARY HYPERALDOSTERONISM
BACKGROUND AND PATHOGENESIS
The first feline case of hyperaldosteronism related to an adrenocortical carcinoma was reported in 1983 involving a 17-year-old domestic shorthair cat presenting with hypokalemic polymyopathy.31 Since 1999 PHA has been reported in 34 more cats. Adrenocortical adenoma or adenocarcinoma was identified in 23 cats.32–41 Of these 23 cats, 13 were included in a single case series36; the series allowed a more detailed profile of this disease to be described. Interestingly, nine of the 13 cats were diagnosed at a single first opinion practice in the United Kingdom, raising the possibility that the condition might be more common than thought previously.
Unilateral adrenocortical neoplasia (adenoma or carcinoma) is the most commonly described cause of PHA in cats.31–41 In contrast to human beings in whom adrenal carcinomas are very rare, unilateral adrenal adenomas and carcinomas appear to have approximately equal incidence in cats. Bilateral adrenal neoplasms also have been described in cats. Interestingly one patient initially had a left adrenal adenoma removed surgically, but by 2 years later the cat developed a right adrenal carcinoma.33 Two cases of bilateral adrenal adenomas have been identified, but for both cases the bilateral nature of adrenal disease was only discovered at postmortem examination, highlighting the necessity for careful evaluation of both adrenal glands.36
Aldosterone-producing adrenal neoplasms also have been recognized in conjunction with other endocrinopathies. Multiple endocrine neoplasia type 1 was reported in one cat with an aldosterone-producing adenoma and concurrent insulinoma and functional parathyroid adenoma.38 One of the authors of this chapter (AH) has observed both hyperthyroidism and PHA in cats. Some adrenocortical tumors, especially carcinomas, may secrete glucocorticoids or sex steroids in addition to mineralocorticoids. Concurrent diabetes mellitus and primary hyperadrenocorticism have been reported.32,37 To date there have not been detailed studies of steroid profiles in cats with PHA, but excessive co-secretion of glucocorticoid or progesterone may explain the occurrence of both hyperaldosteronism and diabetes mellitus.37
In 2005, a series of 11 cats were described as having nontumorous PHA, providing evidence for a feline counterpart of bilateral adrenal hyperplasia in human beings.42 Most of these cats had laboratory evidence of mild renal disease. Three of the 11 cats with bilateral adrenal hyperplasia underwent necropsy with identification of bilateral nodular hyperplasia of the zona glomerulosa. Histopathological changes in the kidneys revealed hyaline arteriolar sclerosis, glomerular sclerosis, tubular atrophy, and interstitial fibrosis. Thus the causes of PHA (adrenal tumor or hyperplasia) that are most common in human beings also occur in cats.
FELINE PRIMARY HYPERALDOSTERONISM: ADRENAL ADENOMA AND CARCINOMA
CLINICAL SIGNS RELATED TO MINERALOCORTICOID EXCESS
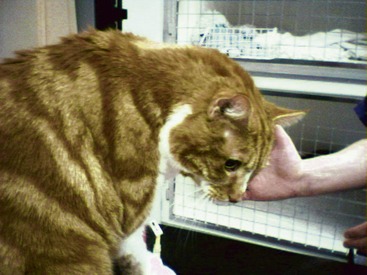
Most commonly the major presenting signs relate directly to increased aldosterone concentrations. The clinical presentation of cats can be divided broadly into two main groups: hypokalemic polymyopathy and acute onset blindness. The former is the most common presentation, with most reported cases presenting with various signs consistent with muscle dysfunction, including cervical ventroflexion (most common; Figure 24-3), hind limb weakness and ataxia, or, less commonly, limb stiffness, dysphagia, and collapse. In some cases the muscular features are mild and episodic, while in others they are severe and acute in onset. One report described a cat presenting with respiratory distress, and the authors speculated that the respiratory failure was a manifestation of hypokalemic polymyopathy.39 The cat was dehydrated upon admission and received intravenous fluids. With rehydration serum potassium became lower, presumably caused by dilution and because increased renal perfusion allowed more potassium loss. This response serves to illustrate that dehydration may mask the severity of hypokalemia or, perhaps, hypertension.
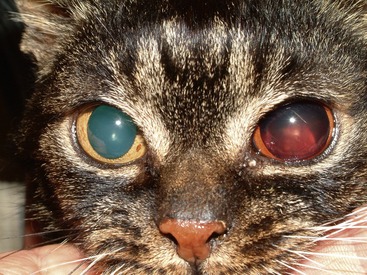
Intraocular hemorrhage or acute onset blindness resulting from retinal detachment (Figures 24-4 and 24-5) is the main presenting sign less commonly, occurring in just two of the reported cases. In both patients, hypertension was severe (systolic blood pressure above 200 mm Hg), and no other clinical signs were reported. However, as stated, although not always the main presenting sign, subclinical hypertension is common.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


