Pharmacotherapy describes the treatment of disease through the administration of drugs.1 In the context of this chapter, drugs are chemical substances used to mitigate disease or otherwise enhance physical or mental wellbeing. This chapter excludes discussion of biologics which are also chemical moieties used to treat disease but more generally composed of sugars, proteins, nucleic acids or more complex combinations of these substances, including cells and tissues.2 Nonsteroidal anti-inflammatory drugs represent a class of drugs that inhibit one or more of the pathways involved with the synthesis of prostaglandins (PGs) and thromboxanes (TBXs) from arachidonic acid. The principal mechanism of action is through inhibition of the enzyme cyclooxygenase (COX). Prostaglandins, particularly those of the E-series, are involved in synovial inflammation and depletion of proteoglycan from articular cartilage matrix.3–4 There is substantial evidence to suggest that PGE2 is the principal PG involved in joint degeneration. Synoviocytes and chondrocytes synthesize PGE2 in response to exposure to lipopolysaccharide (LPS) and other inflammatory mediators, and increased concentrations of PGE2 have been reported in the synovial fluid of horses with osteoarthritis.5–6 Additionally, there is also evidence to suggest that PGs may modulate latent matrix metalloprotease release with resultant cartilage matrix degradation.7–8 Nonsteroidal anti-inflammatory drugs are classified based upon their chemical structure into carboxylic and enolic acids. A majority of NSAIDs belong to the carboxylic acid group, while fewer are enolic acids. Many texts additionally divide NSAIDs into numerous and often confusing sub-categories based, for example, on their chemical properties, mode of action, or pharmacologic effect.9 This text will focus instead on their cyclooxygenase enzyme (COX)-1, -2 and -3 selectivity. Most NSAIDs have a similar mechanism of action as well as physical and chemical properties.10–11 Nonsteroidal anti-inflammatory drugs inhibit the synthesis of prostaglandins (PGs) and thromboxanes (TBXs) from arachidonic acid by inhibiting COX. Most NSAIDs are also competitive COX antagonists; this mechanism of action of NSAIDs requires the continued presence of the active drug to exert the effect. Aspirin, however, is an irreversible antagonist that deactivates COX by acetylation. Because platelets are nonnucleated and thus do not have the capacity to synthesize new COX, ASP has a profound effect on platelet function. There are at least two COX isomers, namely the constitutive (COX-1) found throughout the body and the so-called inducible (COX-2) isoforms.9 The COX-1 isomer is the ‘housekeeping’ enzyme responsible for basal synthesis of PGs necessary for physiologic functions, including maintenance of vasomotor tone (intestinal mucosal and renal blood flow), prevention of platelet aggregation and adhesion and maintenance of gastric cytoprotection.9 The COX-2 isomer is responsible for increased eicosanoid synthesis associated with inflammation and is induced by lipopolysaccaride (LPS), cytokines and other inflammatory mediators. However, the ‘good’ COX-1 versus ‘bad’ COX-2 dichotomy has proved overly simplistic. COX-2 has been shown to exert its own physiological effects, for instance in regulating renal blood flow12 and GI mucosal integrity, especially in ulcer healing.13–14 A third COX isoform, sensitive to acetaminophen and referred to as COX-3, COX 1b or COX-1 variant (because of its similarity to COX-1) has also been identified in some species including the dog.13 The affinity for the COX isoforms varies among NSAIDs. Some are more potent inhibitors of COX-1, while some are relatively equipotent for their effects on COX-1 and COX-2, and others have a greater selectivity for COX-2. The COX-2/COX-1 ratio is a convention used to reflect the relative selectivity of an NSAID for the COX isomers.9 Unfortunately, the convention can be confusing to the reader because the numerator/denominator values (COX-2/COX-1) actually reflect the IC50 for each isomer. The IC50 represents the concentration of a drug that is required for 50% inhibition in vitro.9b Hence, an NSAID with a COX-2/COX-1 ratio <1 is preferable in order to maximize the anti-inflammatory effects of the NSAID while minimizing the potential for toxic side effects. With that in mind, an array of COX-2 specific inhibitors, known as coxibs, have been developed and some have been used in horses. Currently, firocoxib (FCB) is the only COX-2 selective drug labeled for use in horses in the United States, while meloxicam (MLX), which is somewhat selective for COX-2, is approved for use in horses in Europe.15 The concentration of firocoxib required to inhibit 50% of COX-1 activity in horses is reported to be >640 times that required to inhibit 50% of COX-2 activity.16 Furthermore, concentrations of firocoxib that inhibit 80% and 50% of COX-2 activity in vitro (67 and 30 ng/mL, respectively) are readily achievable in equine plasma following oral administration at the label dose of 0.1 mg/kg.17 The relative potency of the COX-2 versus COX-1 drugs as analgesics when administered at their approved dose rates has been the subject of debate. However, in a study that compared the efficacy and safety of FCB (0.1 mg/kg, PO, SID) and PBZ (~4.4 mg/kg, PO, SID) paste formulations administered for 12–16 days to 253 horses with chronic arthritis, the proportion of horses clinically improved on day 14 for the FCB group (104/123 [84.6%]) was not significantly different from the proportion for the PBZ group (103/119 [86.6%].18 In a second study, when FCB was administered as an oral paste (0.1 mg/kg, PO SID) for 14 days to 390 horses afflicted with osteoarthritis, improvement was reported in approximately 80% by day 14.19 Finally, when oral MLX (0.6 mg/kg) was administered to horses with induced intercarpal joint synovitis, lameness and joint effusion was alleviated compared to placebo-treated joints, as were synovial fluid biomarkers (PGE2, substance P, bradykinin, MMP), and some turnover markers of aggrecan (GAG) and type II collagen (CPII, C2C).20 Peak plasma concentrations and the onset of action for NSAIDs after oral administration varies with the timing of administration relative to eating. Mean time to peak plasma concentration for PBZ (4.4 mg/kg PO) in ponies with access to hay was delayed 6 to 12 hours, compared with fasted ponies.21 Phenylbutazone and other NSAIDs appear to bind to hay and other digesta, thus delaying time to peak plasma concentration. Fermentative digestion of roughage in the large intestine releases the bound drug, which possibly contributes to the propensity of NSAIDs to cause ulcerative disease in the large intestine.10 The time to reach peak plasma concentration and the elimination half-life can be dramatically prolonged by the timing of administration relative to feeding. This has important clinical implications with regard to the therapeutic efficacy of an orally-administered NSAID as well as with potential problems related to horses in sanctioned competitive events. There is conflicting data regarding the effects of feeding versus fasting on the absorption of MFA.9,22 Horses with free access to feed had delayed absorption, decreased maximal plasma concentration, and delayed time to peak concentration, compared with fasted horses, administered FLM. Although it may not be practical, NSAIDs should be administered to horses that have not eaten for at least two hours before, and are withheld from feeding for two hours after, dosing in order to control absorption, time to peak plasma concentration, and elimination half-life. Most NSAIDs undergo hepatic metabolism and either renal or biliary excretion. NSAIDs administered to lactating mares are not excreted in milk in concentrations yielding measurable plasma levels in foals. Phenylbutazone undergoes hepatic metabolism to yield oxyphenbutazone, the pharmacologically active metabolite, and γ-hydroxyphenylbutazone, an inactive metabolite. The parent compound and the two metabolites are excreted into the urine, and due to their acidic nature, excretion is more rapid in alkaline than acidic urine.23–24 Aspirin (acetylsalicylic acid) is rapidly deacetylated to salicylate, the active metabolite of the parent drug. A paucity of data regarding metabolism of MFA exists for horses; it is known that conjugation occurs and between 10–14% is excreted in the urine after oral administration.11 Although little is known regarding the elimination of FLM, it does not accumulate in the body and approximately 14% is excreted in urine.11 Naproxen undergoes hepatic metabolism and along with its principal metabolite, 2-(6-hydroxynaphthyl)propionic acid, are excreted in the urine in high concentrations.25 Ketoprofen is used clinically as a racemic mixture of the R(–) and S(+) enantiomers with half-lives of 1.1 hours and 1.5 hours, respectively, after IV administration.26 Ketoprofen accumulates in inflammatory exudates, resulting in substantially prolonged exudate half-lives of 19.7 hours and 22.6 hours for the R(–) and S(+) enatiomers, respectively. Similar to KTP, CRP is used clinically as a racemic mixture of the R(–) and S(+) enantiomers, each with different potencies and pharmacokinetics. There is evidence that the COX inhibitory and anti-inflammatory effects of carprofen are primarily attributable to the S(+) form. However, other evidence suggests that the R(–) enantiomer may be equipotent to the S(+) form as an analgesic and in some other activities such as inhibition of β-glucuronidase release.27–28 Coincident with decreases in plasma concentrations, CRP accumulates in inflammatory exudates resulting in exudate levels exceeding plasma levels from 2 to 48 hours.29 NSAIDs are highly (90 to 99%) bound to plasma proteins, thus, the free active fraction of these drugs is extremely small.9 Concurrent administration of another highly protein-bound drug (eg, chloramphenicol, rifampin, barbiturates) can cause displacement of the NSAID resulting in an increased free active fraction.9 The pharmacokinetics of NSAIDs varies with the age of the horse. The plasma half-life of IV PBZ (4.4 mg.kg) was longer for 8–10 year old adult horses and ponies (5.5 hours) than for three-year-old ponies (3.9 hours)21. NSAIDs have substantially different pharmacokinetic profiles in neonatal (<24 hours of age) foals compared with adult horses. In general, neonates have a reduced ability to eliminate NSAIDs, compared with adult horses, after a single IV dose30–32. The large volumes of distribution of NSAIDS in neonatal foals may necessitate larger initial doses, however, the relatively long half-lives suggests that the dosing interval should be extended to prevent accumulation of these drugs with resulting potential toxic side effects. Unlike most NSAIDS, PBZ can have dose-dependent kinetics, with the half-life increasing from 4 to 8 hours when administered at a dose of 10 mg/kg.33 This effect is likely due to saturation of hepatic enzymes responsible for PBZ metabolism. Taken together, the effect of dose and age on the pharmacokinetics of PBZ emphasizes the need for caution in the dose and dosing interval when administering this drug to older horses, especially those that are systemically ill or dehydrated. Phenylbutazone, like many NSAIDs, has dramatically different kinetic values for tissue compared with plasma.9 The acidic nature and high protein binding of NSAIDs causes them to accumulate at sites of inflammation. For example, the plasma half-life for PBZ is between 4 and 8 hours, whereas the half-life in exudates is approximately 24 hours.34 Ketoprofen accumulates in inflammatory exudates, and thus the half-lives of the R(–) and S(+) enantiomers are prolonged from 1.1 hours and 1.5 hours to 19.7 hours and 22.6 hours, respectively.35 Similarly, CRP accumulates in exudates and yields substantially greater concentrations than in plasma between 2–48 hours after administration.29 Concurrent administration of chloramphenicol reduces the clearance and increases the half-life of PBZ, which may accentuate both therapeutic and toxic effects. A single dose (25 mg/kg IV) of chloramphenicol prior to PBZ administration resulted in a significant decrease in the elimination rate, and this effect was accentuated by additional dosing of chloramphenicol.36 Rifampin induces hepatic biotransformation processes and has been shown to increase the elimination rate of PBZ in horses.36 There are anecdotal reports that when PBZ is administered to horses anesthetized with thiobarbiturates increased depth and duration of anesthesia may occur due to competitive plasma protein binding.37–38 A study evaluating administration of PBZ (8.8 mg/kg IV) nine minutes after thiamylal (11 mg/kg IV) demonstrated no effects of PBZ on thiamylal pharmacokinetic parameters or depth or duration of anesthesia.39 However, there were changes in PBZ pharmacokinetics, including increased serum concentrations and decreased percentage protein bound PBZ. In another study, administration of PBZ (6.6 mg/kg IV) for four days prior to thiamylal anesthesia did not have a significant effect on recumbency time.36 Therefore, it seems unlikely that perianesthetic administration of PBZ would alter the intensity or duration of anesthesia. It also seems unlikely that thiamylal administration would have a clinically important effect on the disposition of PBZ. A study evaluating the pharmacokinetics of steady-state PBZ and single bolus gentamicin administered together revealed that gentamicin pharmacokinetics are altered by concomitant PBZ therapy, however, no effect of gentamicin was found on the pharmacokinetics of PBZ.40 Gentamicin was administered as a 2.2 mg/kg IV bolus on the fourth day of once daily treatment with PBZ (4.4 mg/kg IV). Phenylbutazone induced a 49% increase in the rate of gentamicin return to the central compartment from peripheral tissues and the half-life and volume of distribution of gentamicin decreased 23% and 26%, respectively. There were no changes in PBZ pharmacokinetics induced by gentamicin. Because PBZ induces changes in the rate and extent of distribution and elimination of gentamicin, caution should be exercised when using this drug combination in horses. The dose, interval, and route of administration for NSAIDs commonly used in horses are given in Table 23.1.11,22,29,41–47 Table 23.1 Recommended dosage regimens for nonsteroidal anti-inflammatory drugs in horses† PO = per os, IV = intravenous, IM = intramuscular, SID = once daily, BID = twice daily, TID = three times daily, QID = four times daily, QOD = every other day NSAIDs in combination. Ponies may require lower dosages10. †Dosages are based on normal horses; may need to adjust dosages if horses are ill, dehydrated, volume depleted, or if administering Modified from Kallings P: Nonsteroidal Anti-inflammatory Drugs. Vet Clin North Am: Equine Practice 9: 523–541, 1993, with permission from Elsevier. Although the mechanism of action is the same, there are apparent differences in the efficacy of different NSAIDs depending upon the type of condition being treated. For example, clinical and experimental data suggest PBZ is more efficacious in providing analgesia for most horses with musculoskeletal disease, whereas FLM is more effective in providing visceral analgesia in horses with colic.48 These differences in effect may be due to the specificity of certain NSAIDs for different COX isomers or for COX within different tissues. Ketoprofen is reported to be effective in decreasing inflammation and pain associated with musculoskeletal disease and colic.49–51
Pharmacotherapy of joint and tendon disease
Introduction
Nonsteroidal anti-inflammatory drugs
Classification and mechanism of action
Pharmacokinetics
Absorption
Metabolism and elimination
Plasma protein binding
Effect of age on pharmacokinetics
Effect of dose on pharmacokinetics
Tissue kinetics
Drug interactions
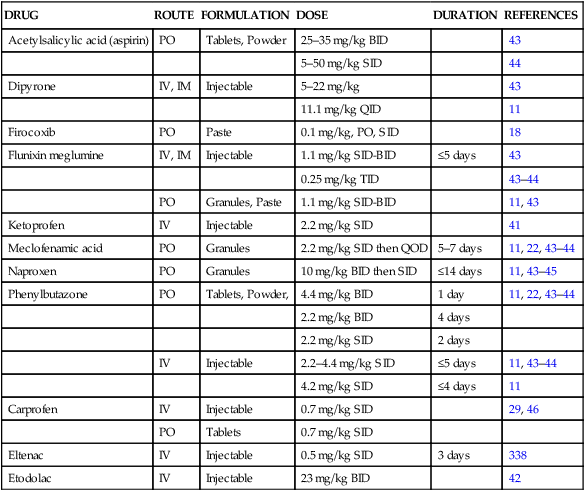
Dosage regimen
DRUG
ROUTE
FORMULATION
DOSE
DURATION
REFERENCES
Acetylsalicylic acid (aspirin)
PO
Tablets, Powder
25–35 mg/kg BID
43
5–50 mg/kg SID
44
Dipyrone
IV, IM
Injectable
5–22 mg/kg
43
11.1 mg/kg QID
11
Firocoxib
PO
Paste
0.1 mg/kg, PO, SID
18
Flunixin meglumine
IV, IM
Injectable
1.1 mg/kg SID-BID
≤5 days
43
0.25 mg/kg TID
43–44
PO
Granules, Paste
1.1 mg/kg SID-BID
11, 43
Ketoprofen
IV
Injectable
2.2 mg/kg SID
41
Meclofenamic acid
PO
Granules
2.2 mg/kg SID then QOD
5–7 days
11, 22, 43–44
Naproxen
PO
Granules
10 mg/kg BID then SID
≤14 days
11, 43–45
Phenylbutazone
PO
Tablets, Powder,
4.4 mg/kg BID
1 day
11, 22, 43–44
2.2 mg/kg BID
4 days
2.2 mg/kg SID
2 days
IV
Injectable
2.2–4.4 mg/kg SID
≤5 days
11, 43–44
4.2 mg/kg SID
≤4 days
11
Carprofen
IV
Injectable
0.7 mg/kg SID
29, 46
PO
Tablets
0.7 mg/kg SID
Eltenac
IV
Injectable
0.5 mg/kg SID
3 days
338
Etodolac
IV
Injectable
23 mg/kg BID
42
Therapeutic uses
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Pharmacotherapy of joint and tendon disease
