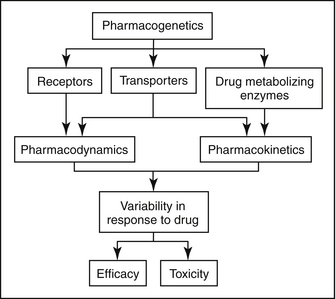
3 Pharmacogenetics
Genetic variation can affect both the pharmacokinetics (i.e., drug absorption, distribution, metabolism, and excretion) and pharmacodynamics (i.e., interaction with drug transporters and receptors) of pharmaceutical agents (Figure 3-1). Currently, the greatest body of knowledge with regard to pharmacogenetics involves genetic variation in drug metabolism. Indeed, the concept of pharmacogenetics originated in the 1950s as a result of the observation in human beings that two populations of individuals existed with respect to their ability to metabolize succinylcholine, followed by the realization that this trait was inherited.1 Approximately 1 in 3500 Caucasian subjects is homozygous for a mutation in the butyrylcholinesterase gene.2 Because this gene is responsible for hydrolysis and inactivation of succinylcholine, patients who have the mutation experience prolonged muscle paralysis and apnea. At the time, molecular biological techniques had not yet been discovered, so the field of pharmacogenetics was initially based purely on phenotypic observations.
Pharmacogenetics of Drug Absorption
Until recently, systemic bioavailability of orally administered drugs was considered to be a function of physicochemical characteristics of the drug and subsequent hepatic metabolism. A number of other factors have recently been shown to affect the ability of a drug to be absorbed into the systemic circulation after oral administration. Intestinal phase I drug metabolism and active drug extrusion by efflux transporters are now considered to be among the most important determinants of oral drug bioavailability. Consequently, polymorphisms in intestinal drug-metabolizing enzymes and drug transporters should dramatically affect oral drug absorption.
In humans CYP 3A enzymes are expressed at higher levels in mature villus tip enterocytes than in hepatocytes.3 Because intestinal villi make up such a large surface area, there is a high likelihood that absorbed drug will interact with intestinal CYP 3A enzyme, facilitating substantial first-pass metabolism. Interpatient variability in intestinal CYP 3A levels has been studied in a small sample of human patients. Elevenfold variations in CYP 3A protein content and sixfold variation in enzymatic activity were identified, suggesting that CYP 3A polymorphisms exist in the human population.4
Intestinal drug metabolism is thought to be important in veterinary patients also, but relatively little is known with regard to interpatient variability in enzyme activity. In dogs tissue distribution (specifically liver and duodenum) of CYP3A12 and CYP3A26 mRNA has been investigated by the author. Overall, expression of CYP3A mRNA was greater in liver than in duodenum, but the relative contributions of the two canine CYP3A isoforms differed. Hepatic expression of CYP3A26 was greater than CYP3A12 in all dogs, with CYP3A26 making up 75.2% of the hepatic CYP3A pool. Conversely, duodenal expression of CYP3A12 was greater than CYP3A26 in all dogs, with CYP3A12 composing 99.8% of the duodenal CYP3A pool.5 Additionally, there was a large amount of interindividual variability among dogs. Further research is needed in this area to determine whether genetic differences account for the variation in expression of this important drug-metabolizing enzyme in hepatic and intestinal tissue among dog breeds and/or individual dogs.
Drug transporters are also known to play an important role in drug absorption. Many drug transporters have been identified in people, but the most well-characterized drug transporter is P-glycoprotein (P-gp), the product of the MDR1 (ABCB1) gene. The potential impact of transporter pharmacogenetics on drug pharmacokinetics is dramatically illustrated by P-gp. P-gp is a transmembrane protein that was first described in highly resistant tumor cell lines.6 Tumor cells expressing P-gp were cross-resistant to various anticancer agents (anthracyclines, vinca alkaloids, taxanes, and others). P-gp has since been shown to act as an ATP-dependent pump that exports drugs from cells. P-gp is also expressed in normal mammalian tissues, where it appears to function in a protective capacity. P-gp is expressed on bile canaliculi, renal tubular epithelial cells, the placenta, brain capillary endothelial cells, and at the luminal border of intestinal epithelial cells.7 At these locations P-gp pumps transported drugs either out of the body (into the bile, urine, or intestinal lumen) or away from protected sites (brain tissue, fetus).
The significant role intestinal P-gp can play in determining oral drug bioavailability has been demonstrated in rodent studies. In mdr1(-/-) knockout mice, oral bioavailability of many P-gp substrate drugs (vinblastine, taxol, digoxin, loperamide, ivermectin, cyclosporine A, others) are substantially greater than in wild-type mice.8,9 Similarly, MDR1 polymorphisms in humans have been shown to result in altered oral bioavailability of P-gp substrate drugs. Studies have shown that oral bioavailability of digoxin, a P-gp substrate, is greater in subjects with the 3435TT MDR1 genotype compared with those with the MDR1 3435CC genotype.10 Similarly, the P-gp substrate phenytoin has been shown to have lower oral bioavailability in subjects with the MDR1 3435 CC genotype.11
P-gp has been fairly well characterized in dogs. Tissue distribution of P-gp in dogs is similar to that in people,12 and it has been shown to contribute to chemotherapeutic drug resistance in vitro and in vivo.13–15 Although its role in determining oral drug bioavailability is not well characterized, there is some evidence that P-gp is important. Bioavailability of the anticancer agent (and P-gp substrate) docetaxel was increased seventeenfold when co-administered with a P-gp inhibitor.
The MDR1 polymorphism in dogs consists of a four base-pair deletion mutation. This deletion results in a shift of the reading frame that generates several premature stop codons.16 Because protein synthesis is terminated before even 10% of the protein product is synthesized, dogs with two mutant alleles exhibit a P-gp null phenotype, similar to mdr1 (-/-) knockout mice. Affected dogs include many herding breeds. For example, roughly 75% of Collies in the United States, France, and Australia have at least one mutant allele.17 Other affected herding breeds, albeit affected at a lower frequency, include Old English Sheepdogs, Australian Shepherds, Shetland Sheepdogs, English Shepherd Dogs, Border Collies, German Shepherd Dogs, Silken Windhounds, McNab Shepherds, and Longhaired Whippets.18
One group of investigators describe a modest effect of the MDR1 deletion mutation on oral drug availability.19 Three P-gp substrate drugs (fexofenadine, loperamide, and quinidine) were simultaneously administered to two groups of dogs, dogs homozygous for the MDR1 deletion mutation (MDR1 mut/mut) and MDR1 wild-type dogs (MDR1 WT/WT). Statistically, there were no differences in area under the curve for any drug between the two groups. Plasma concentrations of fexofenadine were significantly higher at two time points in MDR1(mut/mut) dogs than in MDR1(WT/WT) dogs.19 The investigators suggest that P-gp limits intestinal absorption of fexofenadine, an antihistamine (H1), in dogs.
Pharmacogenetics of Drug Distribution
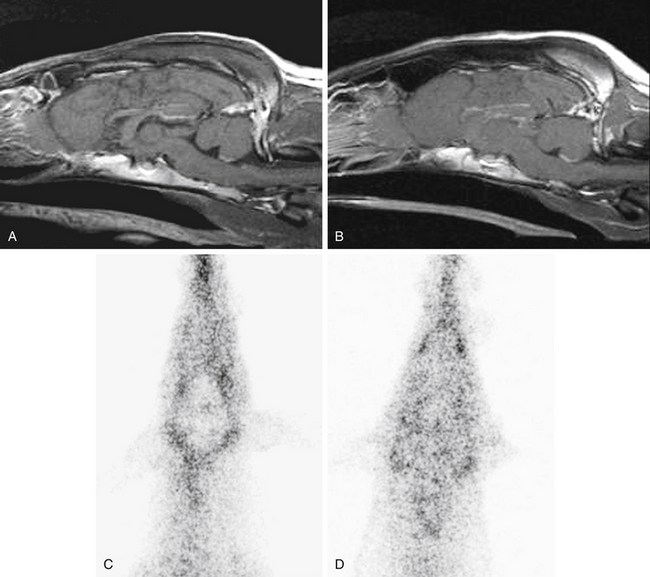
Drug distribution, the delivery of drugs from the systemic circulation to tissues, can be dramatically affected by pharmacogenetics. The drug transporter P-gp serves as an important barrier to the distribution of substrate drugs to selected tissues. For example, P-gp is a component of the blood–brain barrier, the blood–testes barrier, and the placenta. Therefore distribution of P-gp substrate drugs to these tissues is greatly enhanced in MDR1(mut/mut) dogs. MDR1(mut/mut) dogs experience adverse neurologic effects after a single dose of ivermectin (120 μg/kg). Heterozygous [MDR1(WT/mut)] or MDR1(WT/WT) dogs are not sensitive to ivermectin neurotoxicity at the 120 μg/kg dose, but MDR1(WT/mut) animals may experience neurotoxicity at ivermectin doses greater than 120 μg/kg, particularly if daily doses are administered (i.e., protocols for treatment of demodectic mange). Affected dogs also appear to have increased susceptibility to neurologic adverse effects of other avermectins, including milbemycin, selamectin (Revolution package insert), and moxidectin.20 Interestingly, a retrospective study conducted by a national veterinary poison center reported that Collies were overrepresented in canine cases of loperamide-induced neurotoxicity.21 Many Collies displayed signs of neurologic toxicity after administration of routinely recommended doses of the antidiarrheal agent loperamide (Imodium). Loperamide is an opioid that is generally devoid of central nervous system activity because it is excluded from the brain by P-gp.22,23 Loperamide neurotoxicity was recently reported in a Collie that had received a routine dose (0.14 mg/kg orally).24 The dog in this report had the MDR1(mutant/mutant) genotype. MDR1 (WT/WT) dogs do not exhibit neurologic signs after receiving even higher doses of loperamide, indicating that P-gp plays a key role in modulating distribution of substrates such as loperamide to canine brain tissue. The images in Figure 3-2 illustrate the role of P-gp in the blood–brain barrier of dogs.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


