Approval date
Drug name
NDA chemical type b
Indication c
Route of administration
Mar 2004
Iquix (levofloxacin)
3
Treatment of bacterial corneal ulcers
Topical
Jun 2004
Istalol (timolol maleate)
3
Treatment of ↑ IOP due to ocular hypertension or open-angle glaucoma
Topical
Dec 2004
Zylet (loteprednol etabonate + tobramycin)
4
Steroid-responsive inflammation associated with presence or risk of bacterial ocular infection
Topical
Dec 2004
Visionblue (trypan blue)
1
Staining of lens anterior capsule as an aid in ophthalmic surgery
Injection into anterior chamber
Dec 2004
Macugen (pegaptanib sodium)
1
Neovascular (wet) age-related macular degeneration
Intravitreal
Dec 2004
Pataday (olopatadine hydrochloride)
3
Treatment of itching associated with allergic conjunctivitis
Topical
Mar 2005
Bromday (bromfenac sodium)
3
Treatment of postoperative inflammation and pain after cataract extraction
Topical
Apr 2005
Retisert (fluocinolone acetonide)
3
Treatment of chronic noninfectious uveitis of the posterior segment
Intravitreal implant
Aug 2005
Alphagan P (brimonidine tartrate)
3
Treatment of ↑ IOP due to open- angle glaucoma or ocular hypertension
Topical
Aug 2005
Nevanac (nepafenac)
1
Treatment of pain and inflammation associated with cataract surgery
Topical
Jun 2006
Lucentis (ranibizumab)
1 (New biological entity)
Neovascular (wet) age-related macular degeneration
Intravitreal
Macular edema following retinal vein occlusion
Sep 2006
Travatan Z (travoprost)
5
Treatment of ↑ IOP due to open-angle glaucoma or ocular hypertension
Topical
Dec 2006
Alaway (ketotifen fumarate)
5
Temporary relief of itching due to allergic/irritant conjunctivitis
Topical
Apr 2007
Azasite (azithromycin)
3
Bacterial conjunctivitis
Topical
Oct 2007
Combigan (brimonidine tartrate + timolol maleate)
4
Adjunctive or replacement treatment of ↑ IOP due to glaucoma or ocular hypertension
Topical
Nov 2007
Triesence (triamcinolone acetonide)
3, 6
Treatment of sympathetic ophthalmia, temporal arteritis, uveitis, ocular inflammation unresponsive to topical corticosteroids, and visualization during vitrectomy
Intravitreal
Jun 2008
Trivaris (triamcinolone acetonide)
3
Sympathetic ophthalmia, temporal arteritis, uveitis, and ocular inflammation unresponsive to topical corticosteroids
Intravitreal
Jun 2008
Durezol (difluprednate)
1
Treatment of inflammation and pain associated with ocular surgery
Topical
Jul 2008
Navstel (hypromellose, dextrose, glutathione, CaCl, MgCl, KCl, NaHCO3 NaCl, Na3PO4)
3
Intraocular irrigating solution for use during surgical procedures
Intraocular
Oct 2008
Akten (lidocaine hydrochloride)
5
Ocular surface anesthesia
Topical
Feb 2009
Tobradex ST (tobramycin + dexamethasone)
3
Steroid-responsive inflammation associated with presence or risk of bacterial ocular infection
Topical
Feb 2009
Membraneblue (trypan blue)
5
Ocular staining to aid in ophthalmic posterior surgery and facilitating removal of epiretinal tissue
Injection – epiretinal tissues or intravitreal
May 2009
Besivance (besifloxacin hydrochloride)
1
Treatment of bacterial conjunctivitis
Topical
Jun 2009
Ozurdex (dexamethasone)
3
Treatment of macular edema following retinal venous occlusion
Intravitreal injection
Treatment of noninfectious uveitis of the posterior segment
Jul 2009
Acuvail (ketorolac tromethamine)
5
Treatment of pain and inflammation after cataract surgery
Topical
Sep 2009
Bepreve (bepotastine besilate)
1
Treatment of itching associated with allergic conjunctivitis
Topical
Sep 2009
Zirgan (ganciclovir)
3
Treatment of acute herpetic keratitis (dendritic ulcers)
Topical
May 2010
Zymaxid (gatifloxacin)
5
Treatment of bacterial conjunctivitis
Topical
Jun 2010
Isopto Carpine (pilocarpine hydrochloride)
3
Treatment of ↑ IOP due to open-angle glaucoma and ocular hypertension
Topical
Management of acute angle-closure glaucoma
Prevention of postoperative ↑ IOP associated with laser surgery
Induction of miosis
Jul 2010
Lastacaft (alcaftadine)
1
Prevention of itching associated with allergic conjunctivitis
Topical
Aug 2010
Lumigan (bimatoprost)
5
Treatment of ↑ IOP due to open-angle glaucoma and ocular hypertension
Topical
Nov 2010
Moxeza (moxifloxacin)
3
Treatment of bacterial conjunctivitis
Topical
Apr 2011
Lotemax (loteprednol etabonate)
3
Treatment of ocular postoperative inflammation and pain
Topical
As shown in Fig. 7.1, most of the recent FDA approvals, almost 50%, were for reformulations of previously approved drugs as opposed to NMEs.
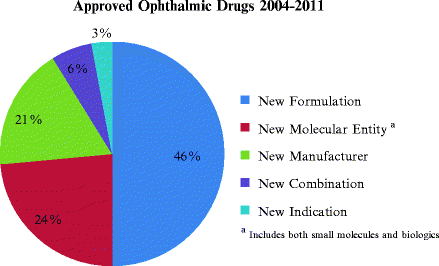
Fig. 7.1
Approved ophthalmic drugs from 2004 to 2011. A total of 33 drugs were reviewed and categorized according to NDA chemical type
A number of clinical trials for therapeutics to treat ocular conditions are ongoing. In April 2011, ClinicalTrials.gov, a website offering up-to-date information on federally and privately supported clinical trials, listed clinical trials for approximately 150 ocular diseases. A partial list of clinical trials that were open in April 2011 is shown in Table 7.2. The clinical trials include those that are recruiting participants, will be doing so in the future, or are evaluating drugs available for individuals with a serious disease who cannot participate in clinical trials (e.g., a patient who might not live sufficiently close to a clinical trial site). The clinical trials include new entities that have not yet been approved as well as approved drugs.
Table 7.2
Open clinical trials for drugs intended to treat ocular conditions (small molecules and biologics)
Indication | Number of clinical trials |
|---|---|
Conjunctival diseases | 72 |
Diabetic retinopathy | 70 |
Dry eye syndrome | 36 |
Macular degeneration | 147 |
Ocular hypertension | 121 |
In the USA, regulatory expectations for the nonclinical studies needed to support the safety of ocular products are not yet defined in a dedicated guidance document. Similar to other drugs, however, therapeutics administered directly on or into the eye undergo safety evaluation in nonclinical studies to support clinical trials and drug approval. Reports from the nonclinical studies are submitted to the US FDA, initially as part of Investigational New Drug (IND) applications to allow the conduct of clinical trials, then ultimately as part of NDAs or Biologics License Applications (BLA) to gain approval for marketing. Also similar to drugs for other indications, drugs for ophthalmic application include new molecular/new biologic entities as well as reformulations of drugs that have already been approved for both ocular and other routes of administration. The nonclinical testing strategy for an ocular drug, however, will be impacted by a number of indication or drug-specific factors, including route of administration (e.g., topical, intravitreal) and extent of systemic exposure. The route- and indication-specific factors result in nonclinical programs for ocular drugs that variably differ from the programs for compounds administered orally or via other systemic routes. Additionally, the ocular product class itself can influence the type of nonclinical program considered appropriate to support safety. Specifically, a more robust nonclinical program might be appropriate for a class of compounds with a novel mechanism of action vs. compounds for which the mechanism of action has been well characterized.
This chapter provides a regulatory perspective on the types of nonclinical safety and toxicity studies needed to support development of ocular therapeutics. This chapter focuses on FDA/Center for Drug Evaluation and Research’s (CDER) regulatory expectations for the nonclinical development of ophthalmic drugs that are administered via an ocular route, such as topical application or intravitreal injection.
7.2 Overview of the Drug Development Process
In the USA as well as in other countries, it is illegal to test new drugs in or to market new drugs to humans without prior approval from the US FDA or the appropriate regulatory body. In order to support the development and approval of drugs, including drugs to treat ocular disease, companies need to present data to the FDA to demonstrate the safety and efficacy of the products being investigated. Generating and reviewing the data required to develop a drug is a multidisciplinary process from both the company and FDA perspective. Drug development involves input from experts in clinical trial design and data evaluation, chemistry and manufacturing, biostatistics, microbiology, and pharmacology/toxicology.
Multiple drug candidates are screened using various assays in the discovery phase of drug development, from which the lead candidate is selected for further development. Nonclinical pharmacology and toxicology studies are needed to support the initiation of clinical trials with the identified drug candidate, and additional nonclinical studies are conducted throughout the drug development process to support the various clinical development phases. Clinical trials are divided into three basic phases as defined in regulations [1], specifically Phases 1, 2, and 3 (Table 7.3).
Table 7.3
Phases of clinical trials
Phase 1 | Phase 2 | Phase 3 |
|---|---|---|
Closely monitored | Closely monitored | Intended to gather information relating to efficacy and safety to allow characterization of risk vs. benefit |
Conducted in patients or normal volunteers | Evaluate the effectiveness of a drug for a given indication(s) in patients with the disease | |
Determine pharmacokinetics, metabolism and/or pharmacology, and potential side effects of escalating doses and, if possible, obtain early evidence of efficacy | Establish short-term side effects and risks | Several hundred to several thousand subjects |
Generally 20–80 subjects | Usually no more than several hundred subjects |
In order to initiate a Phase 1 clinical trial, sponsors (i.e., the companies developing drugs) submit an IND to the FDA. The INDs contain data from the appropriate nonclinical studies, a protocol for the proposed clinical trial, information defining the manufacture of the drug, as well as other pertinent information. If the FDA agrees that the data outlined in the IND support the safety of the proposed clinical trial, sponsors can initiate their clinical program. If the FDA does not agree that the IND adequately supports safety, the company’s program is placed on clinical hold (i.e., clinical trials cannot proceed in the USA) until the safety concern is resolved to the satisfaction of the FDA. As clinical trials progress, sponsors continue to submit nonclinical studies and other information to FDA for review and comment. Once the sponsor has completed all of the appropriate studies to support approval of their drug for marketing, they submit an NDA or BLA for small molecules and biologics, respectively (small molecules vs. biologics are addressed in Sect. 7.6). The properties of approved drugs, including indication, dosing regimen, and potential adverse effects, are described in the approved package insert, which is also referred to as labeling. Nonclinical data, primarily those obtained from genotoxicity, reproductive toxicology, and carcinogenicity studies, are included in the labeling.
The different types of nonclinical studies and their timing within the drug development process are defined in Sect. 7.7. As noted, sponsors need to present nonclinical and clinical data as well as chemistry and manufacturing data to the FDA to support the safety and efficacy of the products being investigated. A team of reviewers at FDA/CDER evaluates the data contained in the INDs and NDAs/BLAs. The team members include the following: medical officers (i.e., clinicians with appropriate expertise in the given indication), nonclinical pharmacology/toxicology reviewers, chemists, clinical pharmacologists, microbiologists, and biostatisticians. The nonclinical pharmacology/toxicology reviewers generally hold PhDs in pharmacology, toxicology, or another life science.
It is possible for sponsors to meet with the FDA throughout the drug development process [2]. The types of FDA meetings, which are identified as Types A, B, and C, are described in Table 7.4. Sponsors submit formal requests for each of the meetings. If the meeting is granted by the FDA, sponsors will provide the FDA with a meeting package outlining the intent of the meeting, the questions that the sponsor is requesting the FDA to address, and the appropriate background information and relevant data. During the pre-IND meeting, sponsors can present their IND-enabling plan (i.e., nonclinical studies conducted to support the safety of clinical trial[s] proposed in the initial submission) with FDA. During the end-of-Phase 2 meeting, sponsors can review completed nonclinical studies and present their plan to support Phase 3 clinical trials and marketing. At the pre-NDA/BLA meeting, the sponsor should confirm with FDA that there are no gaps in their nonclinical program.
Table 7.4
Meetings with FDA
Type A | Dispute resolution meetings |
Meetings to discuss clinical holds | |
Special protocol assessment meetings | |
Should be scheduled within 30 days of FDA’s receipt for request | |
Type B | Pre-IND meeting |
Certain end-of-phase 1 meetings | |
End-of-phase 2 meetings | |
Pre-NDA/BLA meeting | |
Should be scheduled within 60 days of FDA’s receipt of a request | |
Type C | Any meeting other than type A or B |
Should be scheduled to occur within 75 days of FDA’s receipt of a request |
7.3 Regulation of Ophthalmic Products
In the USA, the FDA is responsible for the regulation of all classes of ophthalmic drugs. Additionally, FDA is responsible for the regulation of devices that are being used to deliver ophthalmic drugs. The centers, offices, and divisions within FDA that are responsible for the regulation of ophthalmic products are depicted in Fig. 7.2.
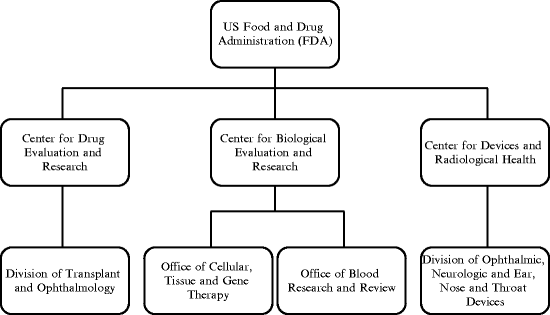
Fig. 7.2
The centers, offices, and divisions within FDA responsible for the regulation of ophthalmic products
The FDA/CDER is responsible for the regulation of the majority of the ophthalmic drug products. The scope of the drugs that FDA/CDER regulates encompasses small molecules, biologics, synthetic peptides, and oligonucleotides. The biologics are comprised primarily of monoclonal antibodies and their fragments, as well as recombinant human proteins such as cytokines and fusion proteins. Like many other organizations, FDA/CDER undergoes periodic reorganizations. Due to workload and other considerations, the ophthalmic drug products have been combined into a division with other drug classes (based on indication) as opposed to being in their own division. Since approximately 1990, the ophthalmic drugs have been paired with five different groups. Although the clinical ophthalmic review staff generally remains the same with the reorganizations, there are often changes in the nonclinical reviewers. Consequently, there may be a loss of consistency with regard to nonclinical expectations and review practices for the ophthalmic drug products.
Pharmaceutical and biopharmaceutical companies are actively pursuing drug candidates from the various drug classifications (e.g., small molecules, biologics, devices) for the treatment of ocular diseases ranging from conjunctival and corneal disorders to retinal diseases. In order to facilitate delivery of ophthalmic drug products, especially to the posterior chamber of the eye, companies are developing devices and other novel delivery systems or are forming partnerships with companies capable of developing delivery systems. As noted, devices are regulated by the FDA Center for Devices and Radiological Health (CDRH). If the therapeutic product consists of a drug that utilizes a device for delivery, the product may be designated as a drug-device combination. The regulatory review of drug-device combinations often involves interaction between CDER and CDRH [3]. If the principle therapeutic moiety is the drug component, however, CDER will typically be the lead center evaluating the product. Combination products are discussed in greater detail in Sect. 7.8.2.
7.4 Nonclinical Guidance Documents and Good Laboratory Practice Regulation
The FDA/CDER’s expectations for each of the review disciplines referenced in Sect. 7.2 are defined in guidance documents. Guidance documents can be grouped into two basic categories, those generated within FDA/CDER and those generated under the International Conference on Harmonisation (ICH) process. Both the FDA/CDER and ICH guidances can be found on the FDA website at http://www.fda.gov/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/default.htm. The FDA/CDER nonclinical guidance documents generally are prepared by a working group consisting of the nonclinical pharmacology and toxicology reviewers within CDER. A partial list of FDA guidance documents is provided in Table 7.5.
Table 7.5
Selected FDA guidance documents pertaining to nonclinical testing to support clinical drug development
Selected FDA nonclinical guidance documents |
|---|
Estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy subjects (2005) |
Genotoxic and carcinogenic impurities in drug substances and products: recommended approaches (Draft, 2008) |
Immunotoxicology evaluation of investigational new drugs (2002) |
Nonclinical safety evaluation of drug or biologic combinations (2006) |
Nonclinical safety evaluation of reformulated drug products and products intended for administration by an alternate route (Draft, 2008) |
The ICH process involves an international cooperative effort to globally harmonize regulatory expectations for drug development. A list of ICH guidances pertaining to nonclinical development is listed in Table 7.6.
Table 7.6
ICH guidance pertaining to nonclinical testing to support clinical drug development
ICH guidance documents |
|---|
ICH S1A; The need for long-term rodent carcinogenicity studies of pharmaceuticals (1996) |
ICH S1B; Testing for carcinogenicity of pharmaceuticals (1998) |
ICH S1C(R2); Dose selection for carcinogenicity studies of pharmaceuticals (2008) |
ICH S2(R1); Genotoxicity testing and data interpretation for pharmaceuticals intended for human use (2011) |
ICH S3A; Toxicokinetics: the assessment of systemic exposure in toxicity studies (1995) |
ICH S3B; Pharmacokinetics: guidance for repeated dose tissue distribution studies (1995) |
ICH S4A; Duration of chronic toxicity testing in animals (1999) |
ICH S5A; Detection of toxicity to reproduction for medicinal products (1994)a |
ICH S5B; Detection of toxicity to reproduction for medicinal products: addendum on toxicity to male fertility (1996)a |
ICH S6(R1); Addendum: preclinical safety evaluation of biotechnology-derived pharmaceuticals (2011) |
ICH S7A; Safety pharmacology studies for human pharmaceuticals (2001) |
ICH S7B; Nonclinical evaluation of the potential for delayed ventricular repolarization (QT interval prolongation) by human pharmaceuticals (2005) |
ICH S8; Immunotoxicity studies for human pharmaceuticals (2006) |
ICH S9; Nonclinical evaluation for anticancer pharmaceuticals (2010) |
ICH M3(R2); Nonclinical safety studies for the conduct of human clinical trials and marketing authorization for pharmaceuticals (2010) |
Nonclinical studies intended to support the safety of clinical trials, such as general toxicology and safety pharmacology studies, need to be conducted in compliance with good laboratory practice (GLP). Primary pharmacology and pharmacokinetic (PK) studies, including absorption, distribution, metabolism, and excretion (ADME) studies, are not required to be conducted according to GLP. It is not uncommon, however, for PK and ADME studies to be conducted in accordance with GLP regulations. Requirements for meeting GLP compliance are defined in the Code of Federal Regulations, 21 CFR 58 [4]. The GLP requirement pertains to all aspects of a nonclinical toxicology study. For example, it requires the following:
1.
That there is a study director who has overall responsibility for the conduct of the study
2.
That there is a quality assurance unit at the testing facility to monitor each study
3.
That there is an analytical method for determining the concentration of test article in the dosing solution(s) used to treat the animals
Contract research organizations (CROs) that conduct nonclinical studies to support drug development and pharmaceutical and biopharmaceutical companies conducting such studies themselves should be capable of conducting studies in compliance with GLP. Because a detailed discussion of GLP compliance is beyond the scope of this chapter, readers are encouraged to refer to the cited CFR for additional information.
7.5 Types of Drug Products
Drug products that are either under development or approved can be divided into the following different categories: NMEs, combination drug products, and reformulations. Before considering the various categories in more detail, it is important to define the difference between drug substance and drug product. The former term refers to the unformulated active ingredient, while the latter refers to the complete dosage form. In addition to the drug substance, the drug product contains excipients that are typically inert substances used to create a suitable clinical formulation for the drug. Excipients include fillers, extenders, diluents, solvents, emulsifiers, preservatives, flavors, absorption enhancers, sustained-release matrices, and coloring agents [5].
An NME is defined as an active ingredient that has never been marketed in the USA in any form. If the NME is a chemically synthesized small molecule, it is referred to as a new chemical entity (NCE). On the other hand, if the NME is a biotechnology-derived protein, such as a monoclonal antibody, it is referred to as a new biological entity (NBE).
As drugs are undergoing development or after they are approved, sponsors might choose to further develop the drug as a combination product. Combination products can be subdivided into three categories: fixed-dose combination (FDC), co-packaged, and adjunctive therapies [6]. The combination product designations are defined in Table 7.7. Nonclinical considerations for combination ocular products are provided in Sect. 7.8.2.
Table 7.7
Definition of combination product designation
Fixed-dose combination | Two or more separate active ingredients combined in a single-dosage form |
Co-packaged | Two or more separate drug products packaged together in their final dosage form |
Adjunctive therapy | A patient is maintained on a second drug product that is used in conjunction with the primary treatment. Relative doses are not fixed, and the different drugs or biologics may or may not be given at the same time. They may be co-packaged |
In addition to developing combination products, sponsors might choose to reformulate a drug product. Reformulation may or may not be associated with a change in route of administration. Generally, additional nonclinical studies are needed to address a change in formulation, especially if it is associated with a change in route of administration. Nonclinical considerations for reformulated ocular drug products are addressed in FDA Guidance for Industry and Review Staff, Nonclinical Safety Evaluation of Reformulated Drug Products and Products Intended for Administration by an Alternate Route [7] and discussed in Sect. 7.8.1.
Both drug substances and drug products can contain impurities. Impurities are defined as any component that is not the drug substance or an excipient in the drug product. They can include by-products, starting materials, degradants, reagents, ligands, and catalysts. The FDA/CDER nonclinical reviewers work closely with the chemistry reviewers in the center to ensure that the safety of impurities is adequately evaluated. Impurities in the drug substance and drug product, including residual solvents, are addressed in ICH guidance documents [8–10]. In addition, the draft FDA guidance, Genotoxic and Carcinogenic Impurities in Drug Substances and Products: Recommended Approaches, addresses a specific concern with respect to impurities [11].
7.6 Biologics vs. Small Molecules and Species Selection
Before considering the types of nonclinical studies needed to support the development of the ophthalmic drugs, it is important to consider differences between chemically synthesized small molecules and biologics. Biologics have properties that distinguish them from small molecules and influence their nonclinical testing strategy. A detailed discussion of the nonclinical development of biologics is beyond the scope of this chapter but is provided in the ICH S6(R1), Preclinical Safety Evaluation of Biotechnology-derived Pharmaceuticals [12]. Additionally, a detailed discussion relating to multiple aspects of biologics development can be found in the text entitled Preclinical Safety Evaluation of Biopharmaceuticals [13]. A summary of the key differences between biologics and small molecules is provided in Table 7.8.
Table 7.8
Comparison of small molecules vs. biologics
Small molecules | Biologics |
|---|---|
Chemically synthesized organic molecule | Proteins obtained from living cells |
Greater potential for off-target effects due to the potential for chemical impurities, active/reactive metabolites, extensive distribution in the body, and activity at multiple receptors or enzymes | Highly targeted, due to lack of chemical impurities and active/reactive metabolites and decreased potential for extensive distribution as a result of large molecular weight |
Generally active and, therefore, potentially toxic in many species | Activity and toxicity generally limited to animals possessing the intended receptor or epitope (i.e., pharmacologically relevant animal model/species) |
Pharmacokinetic and pharmacologic considerations when selecting species for nonclinical studies | Primarily pharmacologic considerations when selecting relevant species for nonclinical studies |
Generally no or negligible potential for immunogenicity | Animals can frequently mount an immune response (immunogenicity) to biologics |
Nonclinical studies for both small molecules and biologics should be conducted in species that are relevant for extrapolation to humans. Relevance can be established based on pharmacology, metabolism, and anatomical considerations. Pharmacological relevance, which refers to a drug’s ability to bind to the intended receptor or other target and elicit the intended pharmacological effect, tends to be more of an issue with biologics but may apply to small molecules as well. The ICH guidance for biologics, ICH S6(R1), clearly states that toxicology studies in nonrelevant species can be misleading and are discouraged [12]. In the case of chemically synthesized small molecules, relevance can be defined, at least in part, by metabolism. A cross-species metabolic stability test that includes metabolic profiling should be conducted using an appropriate in vitro system, such as isolated hepatocytes from humans and laboratory animals, prior to selecting species for toxicology studies of a small molecule.
Ocular anatomical considerations of the various species used in toxicology studies, which may factor into species selection, are addressed in detail in Chap. 1 of this text. With few exceptions, ocular toxicology studies are conducted in nonrodents (e.g., rabbits, dogs, monkeys, and pigs) due to eye size and other anatomical considerations. Although the eyes of rabbits and dogs differ from those of humans, they are routinely used for ocular toxicology studies. Monkey eyes most closely resemble the human eye, and monkeys are often considered the most appropriate species for intravitreal and other treatments administered to the posterior segment of the eye. They are, however, used less frequently than rabbits or dogs due, at least in part, to cost and ethical issues.
Systemic bioavailability refers to the amount of drug absorbed into the systemic circulation following oral or parenteral administration relative to that achieved following intravenous (iv) administration. While bioavailability does not truly define relevance per se, it can be used to select the most appropriate species especially for orally administered compounds. It is generally only a minor consideration for ophthalmic drugs.
7.7 Types of Nonclinical Studies Needed for New Molecular Entities Intended for Ophthalmic Indications
Pharmacology and toxicology studies, which are conducted in laboratory animals and/or in vitro systems, are frequently referred to as nonclinical or preclinical to distinguish them from the clinical trials conducted in humans. Even though selected sections of some of the available guidance documents apply to ophthalmic drugs, there are no published guidance documents dedicated to ophthalmic drugs per se. In 1998, FDA/CDER’s Division of Anti-inflammatory, Analgesic, and Ophthalmic Drug Products presented a poster at the Society of Toxicology meeting on nonclinical development of ophthalmic drug products, which focused on drugs applied topically to the eye. The poster presentation was followed in 1999 by one that addressed intravitreal drug products. Although the abstracts and handouts from these sessions are dated, sponsors still use the documents to some extent as references for the nonclinical development of ophthalmic drug products. Since 1999, FDA/CDER’s pharmacologists/toxicologists have presented their expectations at various scientific meetings, such as Society of Toxicology and other venues, but they have not yet published a guidance document on the topic.
< div class='tao-gold-member'>
Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


