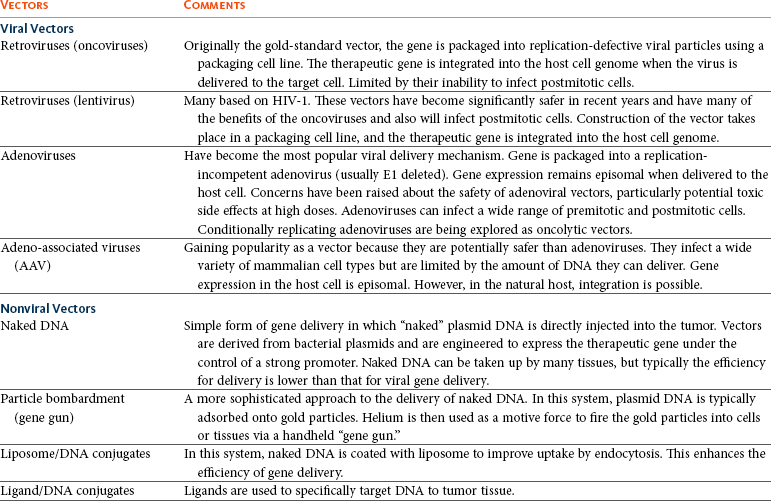
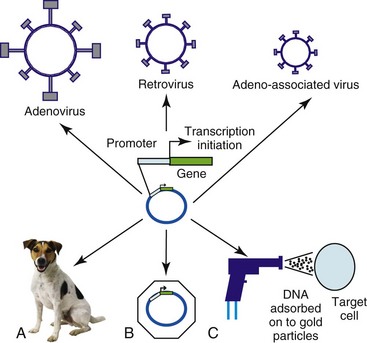
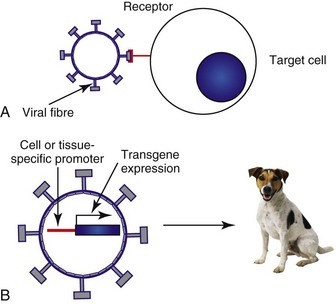
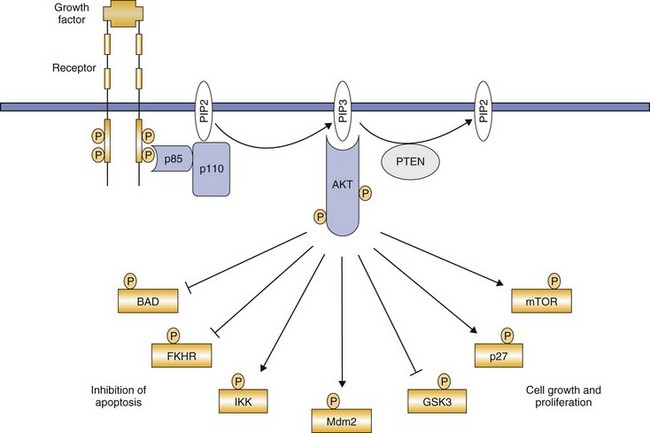
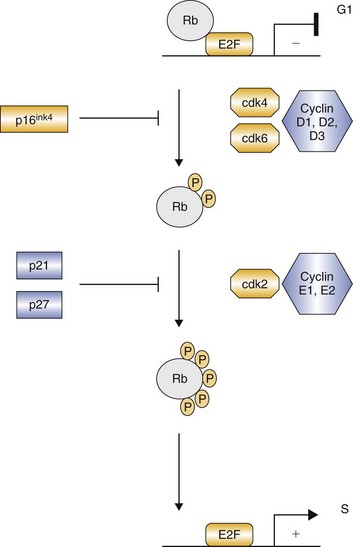
14 Since its development, recombinant DNA technology has been vigorously applied to the advancement of medicine. New molecular techniques have been used to study the role of specific genes and their products in disease, to improve diagnosis, and to produce novel therapeutics. Gene therapy, in its simplest definition, is the introduction of genes into cells in vivo to treat a disease.1 Although one may recognize this as one of the newest areas of medicine, the actual concept of gene therapy is not a new idea. In the late 1960s, the idea of gene therapy was hypothesized by many working in the field of molecular biology, in particular the use of gene therapy to deliver a normal copy of a gene into a patient with a single gene defect, such as hemophilia. However, the technology to manipulate genes and be able to deliver them safely to patients was not available until very recently. Even now and despite improvements in technologies involving gene manipulation and delivery, there are still many technical hurdles to overcome before gene therapy can become accepted clinical practice.1,2 In simple terms, gene therapy is the introduction of nucleic acid into a cell to ameliorate a disease process. For this to be effective, the gene has to be delivered to sufficient numbers of target cells in the body, and this requires a vehicle or vector for delivery. In addition, the gene has to be expressed at a sufficient level and for a length of time appropriate for the disease.2 Early studies on how viruses were able to cause tumors by delivery of their own DNA to foreign cells made them ideal candidates for vectors for gene therapy (i.e., the vehicles by which we could transfer therapeutic genes to patients). However, it was not until the 1980s when work on the retroviral life cycle started to revolutionize the development of gene therapy. In these early studies, it was demonstrated that retroviruses could transfer DNA to cells and this DNA could be stably integrated into the host cell’s genome. Since these early experiments, a number of viruses have been manipulated to act as vehicles for gene delivery. In addition, because of concerns over safety of viral vectors for gene delivery, a number of workers have also explored the possibility of using naked DNA (a therapeutic gene delivered in a bacterial plasmid or lipid/DNA complexes) with some success. The vehicles used for gene delivery and their associated problems are shown in Table 14-1. The ideal vector would efficiently deliver the gene of interest (transgene) specifically to the cancer cell. It would be easy and cheap to manufacture and would be nonimmunogenic. Obviously the ideal vector does not exist, but there have been great advances in both viral and nonviral delivery systems.2 The great advantage to viral vectors for gene delivery is their ability to infect cells and our ability to exploit their replicative machinery. The majority of systems utilize replicative-defective viruses to overcome concerns that recombination within the host may lead to the production of wild-type virus with pathogenic potential. The common systems rely on oncogenic retroviruses (e.g., murine leukemia virus [MuLV]) or adenoviruses (e.g., human adenovirus type 5 [AD5]), but great strides are also being made with lentiviral vectors (particularly human immunodeficiency virus-1 [HIV-1]).3–8 Most of the systems described involve the local delivery of virus to tumor deposits (e.g., by intratumoral injection). Systemic delivery is hindered by rapid clearance of viruses from the body by the immune and complement systems. To overcome this, work is in progress to explore cellular delivery of viruses by the systemic route. In this delivery system, viral producer cells are delivered to the patient, and virus production is triggered when the cells reach the tumor. Endothelial cell cultures lend themselves well to this technology because they specifically home in to areas of neoangiogenesis. However, T-cells, macrophages, and dendritic cells are also being explored as potential cell delivery systems. The advantage of this system is that virus could potentially be delivered to metastatic disease, as well as primary tumors.9–13 Recent evidence suggests that mesenchymal stem cells also have homing ability to tumors and could be used to deliver “suicide” genes (see later). Concerns relating to virus safety and an inability to produce high enough viral titers have led to the development of nonviral delivery systems for gene therapy.14 Liposomes have been used to safely and efficiently deliver genes to tumor cells through direct injection.15,16 Further, naked DNA delivery (the delivery of plasmid DNA alone containing the gene of interest) has been shown to be taken up by tumor cells and antigen-presenting cells after simple direct injection. A modification of this is particle-mediated gene delivery using a “gene gun.” In this approach, DNA is adsorbed on to gold particles and fired into tissues under high pressure (using helium as the motive force).17,18 However, the majority of these techniques are still inefficient vehicles for delivery and are not able to be given systemically (Figure 14-1; see Table 14-1). One of the major barriers to gene therapy becoming accepted in clinical practice is the ability to give vectors systemically and to ensure that therapeutic transgenes are not expressed in normal cells. Numerous strategies have been attempted to provide levels of targeting to spare normal tissue. In a later section, we describe the use of conditionally replicating viruses, which is one method of targeting. Surface modification of viruses (transductional targeting) is also being explored. An example of this is the use of modified fibers on the surface of adenoviruses that only allow the virus to enter cells with specific receptors (Figure 14-2). A further targeting strategy is transcriptional targeting once the vector has entered the cell.19–26 Although every gene is represented in every cell of the body, expression of any one gene requires specific transcription factors that may be unique to a particular cell or tissue type. Certain genes have been identified that are expressed in cancer cells but are not expressed in normal cells (e.g., telomerase) or are only expressed in a specific tissue type (e.g. prostate-specific antigen [PSA]). By using the promoter sequences for these genes to drive transgene expression, targeted expression in cancer cells only (e.g., using the promoter for telomerase) or to a specific tissue type (e.g., to the prostate using the promoter for PSA) can be achieved (see Figure 14-2). The increased understanding of the molecular events in cancer has made possible the identification of defective genes involved in the cancer phenotype. One of the most studied genes in cancer development has been the tumor suppressor gene p53. p53 acts as a genomic guardian for the cell; it is switched on when a cell’s DNA is damaged. The product of this gene causes the cell to either stop dividing or become apoptotic (programmed cell death), depending on the degree of damage. In many cancers, this gene is defective; thus damaged cells fail to stop dividing and can accumulate further damaging events, which can allow selection for a malignant phenotype. A number of studies have addressed this by attempting to replace the defective p53 gene with its normal counterpart.27 However, problems associated with this approach include the following: • The inability of our current technology to be able to efficiently deliver a normal p53 gene to every cancer cell in a tumor mass. • Cancer is a multigenetic abnormality, and the delivery of one correct gene to a tumor cell may still not have the desired phenotypic effect. A more promising approach has been to use the lack of a normal functioning p53 gene to target viruses to kill cells. The use of E1b-deleted adenoviruses to specifically cause oncolysis in p53 null cells is described later and has proved successful in some human clinical models.28 p53 mutations in domestic species such as the dog and cat have also been well characterized, and these may provide targets for therapy, particularly for diseases such as canine osteosarcoma (OSA) and feline vaccine-associated sarcomas (FVAS). Typically, the “suicide gene” approach involves the delivery of a gene (usually an enzyme) to cancer cells that has the ability to convert a relatively nontoxic prodrug to an active compound within the cancer cell (gene-directed enzyme prodrug therapy [GDEPT]) (Figure 14-3). At the clinical level, the gene would be delivered to the patient’s tumor and the enzyme activity would be confined to the cancer cells.29 The patient would then be given a prodrug systemically. In the cancer cells, this novel enzyme can convert the prodrug to a more active compound that has the ability to kill the cancer cell (see Figure 14-3). A number of successful approaches have been developed based on this system. For example, the Escherichia coli nitroreductase gene has been used in preclinical models to cause reduction of an inactive prodrug (CB1954, a weak alkylating agent) to promote cell killing in cancer cells.29 However, due to the low efficiency of existing vectors, the success of this therapy will largely depend on the extent of the bystander effect. In this, the activation of the prodrug in the cell causes cell death and also leakage of toxic metabolites to neighboring cells. Consequently, it is estimated that only a small fraction of the cells need receive the gene for there to be a dramatic effect on tumor volume. Further, in mouse models, a distant bystander effect on tumor metastases has been demonstrated that is mediated through the patient’s immune system.22 The in-situ destruction of tumor cells is mediated through necrosis rather than apoptosis, creating an ideal inflammatory environment for the exposure and presentation of tumor antigens to the immune system. This allows the patient’s immune system to recognize tumor metastases and has caused regression in a number of preclinical model systems. These systems have been combined with transcriptionally targeted vectors (described previously) to improve the eventual therapeutic index. The attractiveness of prodrug cancer gene therapy has been described earlier, but it does rely on the ability to specifically target prodrugs to tumors. This can be achieved using transcriptional targeting of vectors (i.e., the use of cell-specific promoters to drive prodrug gene expression). However, this targeting needs an effective and efficient delivery vehicle, and viruses demonstrate some severe deficiencies in this role. The use of stem cells to target tumors can avoid systemic toxicity. Tumor stroma is composed of a variety of cells, including proliferating tumor cells, cancer stem cells (CSCs), tumor fibroblasts, endothelial cells, lymphocytes, and other cells. Many therapeutic strategies are now geared toward killing both tumor cells and CSCs. Prodrug cancer gene therapy driven by mesenchymal stem cells (MSCs) has been suggested as a treatment modality that could achieve this.30 It represents an attractive tool for activating the prodrug directly within the tumor mass, thus avoiding systemic toxicity. In addition, MSCs lack major histocompatibility complex (MHC)-II and show only minimal MHC-I expression.31–33 Thanks to their immunosuppressive properties, allogeneic MSCs can substitute for autologous stem cells in delivering the therapeutic agent in targeted tumor therapy. Stem cell–driven cancer gene therapy is based on the tumor-trophic property of MSCs. Tumor-homing ability of MSCs holds therapeutic advantages compared to vehicles such as proteins, antibodies, nanoparticles, and to some extent viruses. The success of an enzyme-prodrug gene therapy depends on several factors. The catalytic activity of the enzyme encoded by a suicide gene, a suitable prodrug-enzyme combination, ability of the vector to target tumor cells, sufficient transgene expression, and, importantly, the extent of the bystander effect are the main indicators of effective and successful suicide gene therapy. Various combinations of suicide genes, prodrugs, and gene transfer technologies have been investigated in order to find the most suitable and effective system in combating the otherwise incurable tumors. The failures of the present suicide gene therapies were mainly caused by the inability of vectors carrying the suicide gene to reach invasive tumor cells distant from the tumor bulk, as well as inefficient spread of the vectors within the tumor. Therefore the stem cell–based suicide gene therapy based on the inherent and privileged tumor-trophic nature of MSCs holds great potential for moving suicide gene therapy closer to translation (Figure 14-4). The search for an effective cancer vaccine over the past 150 years has led to extensive studies of the immune response of cancer patients. These studies have suggested that cell-mediated immune responses are important components of the antitumor immune response. Cytokines are small glycoprotein molecules that orchestrate the immune response, tissue repair, and hemopoiesis, and it has been demonstrated that the relative amounts of individual cytokines can direct the immune system toward either a mainly humoral or a mainly cell-mediated response. In particular, cytokines such as interleukin-2 (IL-2), interferon-γ (IFN-γ), IL-12, and IL-18 have the ability to promote cell-mediated responses. Further, evidence derived from animal models suggested that local production of cytokines around a tumor mass can lead to production of an antitumor immune response and a reversal of T-cell anergy (nonresponsiveness).34,35 Thus there appears a rationale for using cytokine molecules in cancer patients to improve the antitumor immune response to tumors that present weakly antigenic epitopes or epitopes that evade immune recognition. In the 1980s and 1990s, a number of clinical studies were undertaken using recombinant cytokine proteins to improve the survival of human cancer patients. However, cytokines tend to be autocrine or paracrine in nature and the levels of protein required to demonstrate a biologic effect were often too toxic for the patient to withstand. However, a more promising approach has been to deliver the actual cytokine genes to cancer cells rather than delivery of the protein to the whole patient.36–38 This approach has also been adopted in a number of small-scale veterinary studies, including one on canine malignant melanoma, which used cells to deliver IL-2 to tumors.39 These studies have had encouraging results and warrant further larger scale trials. In the preceding section, we described how the in-situ destruction of tumor cells by enzymes can lead to a distant bystander effect mediated by the immune system. Many trials have now combined this approach with gene-directed immunotherapy. In this approach, cytokine genes, as well as the prodrug-activating gene, are delivered to cancer cells. The conversion of an inactive prodrug by the activating gene leads to destruction of the cancer cells by necrosis. The co-delivery of cytokines that enhance cell-mediated immune responses such as IL-2, IFN-γ, IL-12, and IL-18 enhances the antitumor response and may potentially improve the distant bystander effect against micrometastatic disease.1,2 An alternative approach to gene therapy for cancer involves the delivery of genes to normal cells of the bone marrow to protect them against the cytotoxic effects of conventional chemotherapeutic drugs. In particular, the multidrug resistance (MDR) gene has been cloned and delivered to normal bone marrow cells. When patients are given high doses of chemotherapy, the normal cells with the MDR gene are able to export the toxic drugs across their membranes and reduce potential side effects.40–42 However, this approach does not protect gastrointestinal cells, which limit this approach. Further, there is also a danger that the MDR gene could transfer to malignant cells, rendering them insensitive to the effects of standard drugs. Progress has been made in the development of replication-competent viruses that conditionally replicate in cancer cells.43–48 As an example, the Onyx 015 vector is an E1b-deleted adenovirus that conditionally replicates in cells with a nonfunctional p53 gene. p53 protein has the potential to shut down cell cycling when infected with wild-type adenovirus but is prevented from doing so through the actions of the product of viral E1b. E1b-deficient viruses cannot replicate in normal cells with p53 intact. However, in cells that have no functional p53 protein, viral replication can proceed and cause cell lysis (Figure 14-5). Many other conditionally replicating viruses are being developed that rely on specific cancer cell defects (e.g., Reo viruses that conditionally replicate in cells with intact Ras signalling pathways) or are transcriptionally targeted. p53 mutations in domestic species such as the dog and cat have also been well characterized, and this may provide targets for therapy, particularly for diseases such as canine OSA and feline vaccine-associated sarcomas. In one study, researchers have utilized the osteocalcin promoter for restricting the replication of a canine adenovirus to dog OSA cells.47 This has shown promise in preclinical evaluations and has been shown to yield a therapeutic benefit in vivo. A cautionary note, however, is that the majority of dogs in the United States and Europe are vaccinated against canine adenovirus, and these vectors may not be able to overcome host immunity. Until recently, many gene therapy trials had utilized retroviral vectors for gene delivery. There are many advantages to using retroviruses as outlined in Table 14-1. However, retroviruses are also associated with serious diseases of domestic animals and the use of these in gene therapy poses a risk of insertional mutagenesis or the production of replication-competent viruses during the manufacturing process. Realistically, insertional mutagenesis leading to a malignant transformation is an unlikely event because cancer is a multistep process. In fact, there may be a greater risk of malignant transformation from external beam radiation than from the use of retroviruses to treat cancer. The production of replication-competent retroviruses during the production process would also be unlikely because of the rigorous testing that is required prior to clinical application. Many of these issues are resolving with the development of new generation vectors.1 As an example, in the use of retroviruses and to prevent insertional mutagenesis in normal tissues, one group recently described the use of zinc finger nucleases (engineered DNA-editing enzymes) that allows the insertion of DNA to a site of choice within the genome.49 This adds a further level of safety in a high-risk procedure. 1. Argyle, DJ. Gene therapy in veterinary medicine. Vet Rec. 1999;144:369–376. 2. Harris, J, Sikora, K. Gene therapy in the clinic. (1993). Aspects Med. 1993;14:251–546. 3. Bartosch, B, Cosset, FL. Strategies for retargeted gene delivery using vectors derived from lentiviruses. Curr Gene Ther. 2004;4(4):427–443. 4. Tomanin, R, Scarpa, M. Why do we need new gene therapy viral vectors? Characteristics, limitations and future perspectives of viral vector transduction. Curr Gene Ther. 2004;4(4):357–372. 5. Lachmann, RH. Herpes simplex virus-based vectors. Int J Exp Path. 2004;85(4):177–190. 6. Buning, H, Braun-Falco, M, Hallek, M. Progress in the use of adeno-associated viral vectors for gene therapy. Cells Tissues Organs. 2004;177(3):139–150. 7. Mah, C, Byrne, BJ, Flotte, TR. Virus-based gene delivery systems. Clin Pharmacokinet. 2002;41(12):901–911. 8. Dornburg, R. The history and principles of retroviral vectors. Front Biosci. 2003;8:D818–D835. 9. Culver, KW, Ram, Z, Wallbridge, S, et al. In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors. Science. 1992;256:1550–1552. 10. Gomez Navarro, J, Contreras, JL, Arafat, W, et al. Genetically modified CD34+ cells as cellular vehicles for gene delivery into areas of angiogenesis in a rhesus model. Gene Ther. 2000;7:43–52. [1]. 11. Harrington, K, Alvarez-Vallina, L, Crittenden, M, et al. Cells as vehicles for cancer gene therapy: the missing link between targeted vectors and systemic delivery? Hum Gene Ther. 2002;13(11):1263–1280. 12. Muta, M, Matsumoto, G, Hiruma, K, et al. Study of cancer gene therapy using IL-12-secreting endothelial progenitor cells in a rat solid tumor model. Oncol Rep. 2003;10(6):1765–1769. 13. Pereboeva, L, Komarova, S, Mikheeva, G, et al. Approaches to utilize mesenchymal progenitor cells as cellular vehicles. Stem Cells. 2003;21:389–404. [4]. 14. Annier, AK, Shea, LD. Controlled release systems for DNA delivery. Mol Ther. 2004;10(1):19–26. 15. Tranchant, I, Thompson, B, Nicolazzi, C, et al. Physicochemical optimisation of plasmid delivery by cationic lipids. J Gene Med. 2004;6:S24–S35. 16. Hirko, A, Tang, FX, Hughes, JA. Cationic lipid vectors for plasmid DNA delivery. Curr Med Chem. 2003;10(14):1185–1193. 17. Yang, N, Sun, WH. Gene and non-viral approaches to cancer gene therapy. Nat Med. 1995;1:481–483. 18. Keller, ET, Burkholder, JK, Shi Pugh, TD, et al. In-vivo particle mediated cytokine gene transfer into canine oral mucosa and epidermis. Cancer Gene Therapy. 1996;3:186–191. 19. Dachs, GU, Dougherty, GJ, Stratford, IJ, et al. Targeting gene therapy to cancer. Oncol Res. 1997;9:313–325. 20. Scanlon, KJ. Cancer gene therapy: Challenges and opportunities. Anticancer Res. 2004;24(2A):501–504. 21. Blackwood, L, Onions, DE, Argyle, DJ. The feline thyroglobulin promoter: towards targeted gene therapy of hyperthyroidism. Domest Anim Endocrinol. 2001;20(3):185–201. 22. Vile, RG, Hart, IR. In-vitro and in-vivo targeting of gene expression to melanoma cells. Cancer Res. 1993;53(5):962–967. 23. Gu, R, Fang, BL. Telomerase promoter-driven cancer gene therapy. Cancer Biol Ther. 2003;2(4):S64–S70. 24. Song, JS. Adenovirus-mediated suicide SCLC gene therapy using the increased activity of the hTERT promoter by the MMRE and SV40 enhancer. Biosci Biotechnol Biochem. 2005;69(1):56–62. 25. Helder, MN, Wiseman, GBA, van der Zee, AGJ. Telomerase and telomeres: From basic biology to cancer treatment. Cancer Invest. 2002;20(1):82–101. 26. Fullerton, NE, Boyd, M, Mairs, RJ, et al. Combining a targeted radiotherapy and gene therapy approach for adenocarcinoma of prostate. Prostate Cancer Prostatic Dis. 2004;7(4):355–363. 27. Edelman, J, Edelman, J, Nemunaitis, J. Adenoviral p53 gene therapy in squamous cell cancer of the head and neck region. Curr Opin Mol Ther. 2003;5(6):611–617. 28. Bortolanza, S, Hernandez-Alcoceba, R, Kramer, G, et al. Evaluation of the tumor specificity of a conditionally replicative adenovirus controlled by a modified human core telomerase promoter. Mol Ther. 2004;9:S375. 29. Blackwood, L, O’Shaughnessy, PJ, Reid, SJ, et al. E. coli Nitroreductase/CB1954: In vitro studies into a potential system for feline cancer gene therapy? Vet J. 2001;161:269–279. 30. Cihova, M, Altanerova, V, Altaner, C. Stem cell based cancer gene therapy. Mol Pharma. 2011;8(5):1480–1487. 31. Le Blanc, K. Immunomodulatory effects of fetal and adult mesenchymal stem cells. Cytotherapy. 2003;5:485–489. 32. Koppula, PR, Chelluri, LK, Polisetti, N, et al. Histocompatibility testing of cultivated human bone marrow stromal cells—a promising step towards pre-clinical screening for allogeneic stem cell therapy. Cell Immunol. 2009;259:61–66. 33. Griffin, MD, Ritter, T, Mahon, BP. Immunological aspects of allogeneic mesenchymal stem cell therapies. Hum Gene Ther. 2010;21:1641–1655. 34. Lasek, W, Basak, G, Switaj, T, et al. Complete tumour regressions induced by vaccination with IL-12 gene-transduced tumour cells in combination with IL-15 in a melanoma model in mice. Cancer Immunol Immunother. 2004;53(4):363–372. 35. Yamazaki, M, Straus, FH, Messina, M, et al. Adenovirus-mediated tumor-specific combined gene therapy using Herpes simplex virus thymidine/ganciclovir system and murine interleukin-12 induces effective antitumor activity against medullary thyroid carcinoma. Cancer Gene Ther. 2004;11(1):8–15. 36. Nagayama, Y, Nakao, K, Mizuguchi, H, et al. Enhanced antitumor effect of combined replicative adenovirus and nonreplicative adenovirus expressing interleukin-12 in an immunocompetent mouse model. Gene Ther. 2003;10(16):1400–1403. 37. Liu, YQ, Huang, H, Saxena, A, et al. Intratumoral co-injection of two adenoviral vectors expressing functional interleukin-18 and inducible protein-10, respectively, synergizes to facilitate regression of established tumors. Cancer Gene Therapy. 2002;9(6):533–542. 38. Goto, H, Osaki, T, Nishino, K, et al. Construction and analysis of new vector systems with improved interleukin-18 secretion in a xenogeneic human tumor model. J Immunother. 2002;25:S35–S41. 39. Quintin-Colonna, F, Devauchelle, P, Fradelizi, D, et al. Gene therapy of spontaneous canine melanoma and feline fibrosarcoma by intratumoral administration of histoincompatible cells expressing human interleukin-2. Gene Ther. 1996;3(12):1104–1112. 40. Carpinteiro, A, Peinert, S, Ostertag, W, et al. Genetic protection of repopulating hematopoietic cells with an improved MDR1-retrovirus allows administration of intensified chemotherapy following stem cell transplantation in mice. Int J Cancer. 2002;98(5):785–792. 41. Schiedlmeier, B, Schilz, AJ, Kuhlcke, K, et al. Multidrug resistance 1 gene transfer can confer chemoprotection to human peripheral blood progenitor cells engrafted in immunodeficient mice. Hum Gene Ther. 2002;13(2):233–242. 42. Fairbairn, LJ, Rafferty, JA, Lashford, LS. Engineering drug resistance in human cells. Bone Marrow Transplant. 2000;25:S110–S113. 43. Chiocca, EA, Abbed, KM, Tatter, S, et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol Ther. 2004;10(5):958–966. 44. Post, DE, Fulci, G, Chiocca, EA, et al. Replicative oncolytic herpes simplex viruses in combination cancer therapies. Curr Gene Ther. 2004;4(1):41–51. 45. Shah, AC, Benos, D, Gillespie, GY, et al. Oncolytic viruses: clinical applications as vectors for the treatment of malignant gliomas. J Neuroncol. 2003;65(3):203–226. 46. Dirven, CMF, van Beusechem, VW, Lamfers, MLM, et al. Oncolytic adenoviruses for treatment of brain tumours. Exp Opin Biol Ther. 2002;2(8):943–952. 47. Hemminki, A, Kanerva, A, Kremer, EJ, et al. A canine conditionally replicating adenovirus for evaluating oncolytic virotherapy in a syngeneic animal model. Mol Ther. 2003;7(2):163–173. 48. Zhan, JH, Gao, Y, Wang, WS, et al. Tumor-specific intravenous gene delivery using oncolytic adenoviruses. Cancer Gene Therapy. 2005;12(1):19–25. 49. Lombardo, A, Genovese, P, Beausejour, CM, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25:1298–1306. Protein kinases play critical roles in normal cell signal transduction, acting to tightly regulate critical cellular processes such as growth and differentiation. These proteins work through phosphorylation; that is, they bind adenosine triphosphate (ATP) and use it to add phosphate groups to key residues on themselves (a process called autophosphorylation) and on other molecules, thereby stimulating a downstream signal inside the cell. This process typically occurs in response to external signals generated by growth factors (GFs) or other stimuli that initiate the cascade. Protein kinases are classified as tyrosine kinases (TKs) if they phosphorylate proteins on tyrosine residues or serine/threonine kinases if they phosphorylate proteins on serine and threonine residues. In some cases the kinases perform both functions (i.e., dual function kinases). These kinases can be expressed on the cell surface, in the cytoplasm, and in the nucleus. The human genome encodes approximately 518 kinases, of which 90 are classified as TKs.1 TKs on the cell surface that are activated through binding of GFs are called receptor TKs (RTKs). Of the 90 identified TKs, 58 are known to be RTKs. Each RTK contains an extracellular domain that binds the GF, a transmembrane domain, and a cytoplasmic kinase domain that positively and negatively regulates phosphorylation of the RTK (Figure 14-6).2–4 Most RTKs are monomers on the cell surface and are dimerized through the act of GF binding; this changes the three-dimensional structure of the receptor, permitting ATP to bind and autophosphorylation to occur, resulting in generation of a downstream signal through subsequent binding of adaptor proteins and nonreceptor kinases.2 Dysregulation of RTKs resulting in pathway activation/uncontrolled signaling is known to contribute to several human cancers, and work is ongoing to characterize such abnormalities in canine and feline cancers. Examples of RTKs known to play prominent roles in specific cancers include KIT, MET, epidermal growth factor receptor (EGFR), and anaplastic lymphoma kinase (ALK), which can be activated by overexpression, mutation, and chromosomal translocation.5–9 RKT signaling is critical for regulating typical cell functions and is also an important regulator of angiogenesis, a process known to be essential for continued tumor cell growth. The RTKs involved in angiogenesis include vascular endothelial growth factor receptor (VEGFR), platelet-derived growth factor receptor (PDGFR), fibroblast growth factor receptor (FGFR), and Tie-1 and Tie-2 (receptors for angiopoietin).10–13 VEGFRs are expressed on vascular endothelium, and VEGFR signaling drives endothelial migration and proliferation.10 PDGFR is expressed on stroma and pericytes that are critical for the maintenance of newly formed blood vessels. It also supports angiogenesis by inducing VEGF transcription and secretion.12,13 FGFR is expressed on vascular endothelium and works with VEGFR to promote increased expression of VEGF.12 Tie-1 and Tie-2 are expressed on blood vessels in tumors and are important in the recruitment of pericytes and smooth muscle cells to the newly forming vascular channels.14 Kinases in the cytoplasm act as a bridge, conducting signals generated by RTKs to the nucleus through a series of intermediates that become phosphorylated.15 The cytoplasmic kinases may be directly on the inside of the cell membrane or free in the cytoplasm. With respect to tumor cell biology, two particular cytoplasmic pathways are often dysregulated in a number of cancers. The first includes members of the RAS-RAF-MEK-ERK/p38/JNK families (Figure 14-7).16,17 Most of these are serine/threonine kinases, and their activation leads to ERK phosphorylation, translocation into the nucleus, and subsequent alteration of transcription factors and nuclear kinase activity important for controlling the cell cycle. Examples of dysregulation in human cancers include RAS mutations in lung cancer, colon cancer, and several hematologic malignancies and BRAF mutations in cutaneous melanomas and papillary thyroid carcinomas.18–20 The second cytoplasmic pathway includes phosphatidyl inositol-3 kinase (PI3K) and its associated downstream signal transducers AKT, nuclear factor κB (NFκB), and mTOR, among others (Figure 14-8).21,22 PI3K is activated by RTKs and in turn activates AKT, which alters several additional proteins involved in the regulation of cell survival, cycling, and growth.23 AKT phosphorylates targets that promote apoptosis (BAD, procaspase-9, and Forkhead transcription factors) and activates NFκB, a transcription factor that has antiapoptotic activity.21–23 AKT also phosphorylates other proteins such as mTOR, p21, p27, and glycogen synthase kinase 3 (GSK3). This leads to redistribution of these proteins either in or out of the nucleus, ultimately inhibiting apoptosis while promoting cell cycling.21–23 Abnormalities of PI3K resulting in pathway activation are commonly found in human cancers, including mutations (breast and colorectal cancers and glioblastoma) and gene amplification (gastric, lung, ovarian cancers).24 This pathway may also become dysregulated through loss of activity of PTEN, a phosphatase that normally acts to dephosphorylate AKT and terminate signaling.21,25,26 PTEN mutations and/or decreased PTEN expression are found in several human cancers (e.g., glioblastoma and prostate cancer)24,25 and have been documented in canine cancers (OSA, melanoma).27–29 RTK-induced signaling ultimately influences cellular events by affecting transcription and the proteins that control cell cycling. The cyclins and their kinase partners (cyclin-dependent kinases [CDKs]) act to regulate the progression of cells through various phases of the cell cycle (Figure 14-9).30–32 The cyclins comprise several families. Cyclins D and E control restriction point passage by activating their respective CDKs (CDK4 and CDK6 for cyclin D and CDK2 for cyclin E). Coordinated function of cyclins D and E is required for cells to progress from G1 into S phase (see Figure 14-9). In many cases, RTK-generated signals induce expression of cyclin D, which complexes with CDK4 and CDK6, resulting in phosphorylation of the tumor suppressor Rb, partially repressing its function.31,32 Functional cyclin D/CDK complexes induce transcription of cyclin E, and active cyclin E/CDK complexes further reduce Rb activity through phosphorylation. This in turn initiates the process of DNA replication important for cell cycling. Dysregulation of the cyclins and CDKs is common in human cancers; for example, overexpression of cyclins D and E is often present in breast, pancreas, and head and neck carcinomas.32 Dysfunction of protein kinases is now recognized to be a common event in tumors. Although this has been best characterized in humans, recent data indicate that dog and cat cancers experience similar dysregulation (Table 14-2). Kinases may be dysregulated through a variety of mechanisms, including mutation, overexpression, fusion proteins, or autocrine loops. In the case of mutations, these may result in phosphorylation of the kinase in the absence of an appropriate signal. Such mutations can consist of a single amino acid change through a point mutation, deletion of amino acids, or insertion of amino acids, usually in the form of an internal tandem duplication (ITD). For example, a point mutation occurs in the BRAF gene (V600E, exon 15) in approximately 60% of human cutaneous melanomas.18,33,34 This amino acid change causes a conformation change in BRAF that mimics its activated form, thereby inducing constitutive downstream ERK signaling and abnormal promotion of cell growth and survival.35,36 RAS is another kinase that is dysregulated through point mutation in several hematopoietic neoplasms (multiple myeloma, juvenile chronic myelogenous leukemia [CML], acute myelogenous leukemia [AML], and chronic myelomonocytic leukemia [CMML]) and in lung cancer, colon cancer, and many others.17,37,38 Table 14-2 Receptor Tyrosine Kinases Associated with Cancer Another example of a mutation involves KIT, an RTK that normally is expressed on hematopoietic stem cells, on melanocytes, in the central nervous system, and on mast cells.39 In approximately 25% to 30% of canine grade 2 and grade 3 mast cell tumors (MCTs), mutations consisting of ITDs are found in the juxtamembrane domain of KIT, resulting in constitutive activation in the absence of ligand binding. These mutations are associated with a higher risk of local recurrence and metastasis.40–42 KIT mutations consisting of deletions in the juxtamembrane domain are also found in approximately 50% of human patients with gastrointestinal stromal tumors (GISTs) and are also found in GISTs in dogs.43–45 There are now several well-characterized mutations involving RTKs in human cancers, including FLT3 ITDs in AML,46–49 EGFR point mutations in lung carcinomas,50,51 and PI3K mutations in several types of carcinomas.24 Overexpression of kinases usually involves the RTKs and may result in enhanced response of the cancer cells to normal levels of growth factor; or, if the levels are high enough, the kinase may become activated through spontaneous dimerization in the absence of signal/growth factor. In humans, the RTK human epidermal growth factor receptor 2 (HER2; also known as ErbB2, a member of the EGFR family) is overexpressed in both breast and ovarian carcinomas and often correlates with a more aggressive phenotype.4,52,53 EGFR is also overexpressed in human lung, bladder, cervical, ovarian, renal, and pancreatic cancers, and some tumors have as many as 60 copies of the gene per cell.7,54,55 As with HER2, such overexpression is linked to a worse outcome in affected patients.7 Fusion proteins are generated when a portion of the kinase becomes attached to another gene through chromosomal rearrangement and the normal mechanisms that control protein function are disrupted. One of the best characterized fusion proteins is BCR-ABL, which is found in 90% of patients with CML.56–59 ABL is a cytoplasmic tyrosine kinase that, when fused to BCR, results in dysregulation of ABL, inappropriate activity of the protein, and resultant malignant transformation. Other examples include TEL-PDGFRβ in CMML, FIP1-PDGFRα in hypereosinophilic syndrome with mastocytosis, and (EML4)-ALK in non–small-cell lung cancer (NSCLC).60 Autocrine loops of activation primarily occur when the tumor cell expresses both the RTK and the growth factor; in most cases, one or the other usually is also overexpressed, resulting in constitutive activation of the RTK. Examples include coexpression of transforming growth factor α (TGFα) and EGFR in glioblastoma and squamous cell carcinoma, insulin-like growth factor (IGF) and its ligand, IGF-1R, in breast and colorectal cancer, and VEGF and VEGFR in melanoma.4,61–63 In canine cancers, possible autocrine loops have been documented in OSA (co-expression of MET and its ligand, HGF) and hemangiosarcoma (HSA; co-expression of KIT and its ligand, SCF).64–66 Several antibodies have been developed to target the extracellular domain of RTKs known to be important in a variety of tumors. These antibodies may prevent the growth factor from binding, may promote internalization of the RTK and subsequent degradation, or may induce an immune response against the cancer cell. One of the most successful examples is a humanized MAb called trastuzumab (Herceptin). This antibody targets HER2, which as previously discussed is overexpressed in approximately 30% of breast cancers, as well as other cancers, including prostate cancer, ovarian cancer, and NSCLC.67 In initial clinical trials, trastuzumab treatment of HER2-positive breast cancer resulted in a response rate of approximately 25% in patients with metastatic disease.68 The response rate approached 50% when trastuzumab was combined with chemotherapy.69 When used in the adjuvant setting, multiple studies have demonstrated that trastuzumab markedly improves survival rates of women with HER2-positive disease; trastuzumab is now part of the routine standard of care for this disease.70,71 Other examples of MAbs that have demonstrated significant activity in human cancers include rituximab (Rituxan) that targets CD20 expressed in a number of B-cell lymphomas72,73 and cetuximab (Erbitux) that targets ERBB1/HER1 EGFR known to be overexpressed in several carcinomas.7,67,74 Small molecule inhibitors work primarily by blocking the ATP-binding site of kinases, essentially acting as competitive inhibitors; a smaller number of these inhibitors work by preventing necessary protein-protein interactions (allosteric inhibition).75 In the absence of ATP, the kinase is unable to phosphorylate itself or downstream signaling elements, thereby interrupting a survival/growth signal essential to the tumor cell, ultimately resulting in cell death. As the molecular characterization of tumors has improved, the development and application of small molecule inhibitors have rapidly expanded in human oncology and their use is markedly altering how cancers are managed. Such inhibitors often are easy to synthesize in large quantities, frequently orally bioavailable, and can readily enter cells to bind the intended target. The first small molecule inhibitor to be approved for human use was imatinib (Gleevec), an orally administered drug that binds the ATP pocket of ABL, as well as the RTKs KIT and PDGFRα.76 As previously discussed, BCR-ABL fusion proteins are present in 90% of human patients with CML, making ABL a good target for therapeutic intervention. The application of imatinib to CML has been transformative, with significant biologic activity demonstrated in several clinical trials, resulting in the approval of imatinib for up-front care of affected individuals.77–82 In the chronic phase of CML, imatinib induces a remission rate of close to 95%, and most patients remain in remission for longer than 1 year. Unfortunately, the remission rate is much lower for patients in blast crisis (20% to 50%), often lasting less than 10 months. Resistance to imatinib has been well characterized and is primarily due to the development of mutations in ABL that preclude drug binding, although gene amplification has also been documented.83,84 Imatinib also has clinical activity against human GIST in which 60% to 80% of the tumors have point mutations or deletions in the juxtamembrane domain of KIT, resulting in constitutive activation.85,86 Response rates of 50% to 70% have been reported with imatinib, far better than the 5% response rate seen with standard chemotherapy.87,88 A small number of GISTs have activating mutations in PDGFRα instead of KIT mutations; these patients also respond to imatinib.89 There are now several small molecule inhibitors approved for the treatment of human cancers that possess specific mutations in kinases known to drive tumor growth and survival. A subset of patients with NSCLC have tumors with activating mutations in EGFR that respond to erlotinib (Tarceva) or gefitinib (Iressa), small molecule inhibitors of EGFR.90 Response rates in patients with EGFR mutations can be as high as 80% compared to less than 10% to 20% for those without, demonstrating that efficacy of targeted therapies often depends on the presence of a known activated signaling element. A small number of patients with NSCLC also exhibit activation of the RTK ALK through its fusion to EML4.91 A small molecule inhibitor of ALK, crizotinib (Xalkori), has demonstrated significant activity against lung cancer patients whose tumors express the EML4-ALK translocation. In a recent phase II study, an objective response rate of 56% was achieved in this subset of patients, with another 31% of patients experiencing stable disease.91,92 As expected with a targeted therapeutic that disrupts key cell-signaling events, responses were rapid with most occurring by 8 weeks of treatment. Vemurafenib (Zelboraf) is a small molecule inhibitor of BRAF that has shown significant activity against cutaneous malignant melanomas that possess activating mutations in BRAF. Most patients treated with vemurafenib experienced tumor shrinkage, with close to 50% meeting the criteria for objective response.93 This compares to an objective response rate of only 5% in patients treated with dacarbazine. Inhibition of mTOR has become of interest in several cancers given the activation of the PI3K pathway and the critical role of mTOR in mediating its effects. Rapamycin, a drug used for many years as an immunosuppressive agent, is the prototypical mTOR inhibitor.94,95 Temsirolimus and everolimus, two rapamycin analogs, have already been approved for use in patients with metastatic renal carcinoma and other mTOR inhibitors are currently under investigation for their potential utility in treating soft tissue sarcomas and bone sarcomas.94,95 Flavopiridol, a partly synthetic flavonoid derived from an indigenous plant (rohitukine) found in India, sits in the ATP-binding pocket of CDK2, acting as a competitive inhibitor.89,90 This compound has been shown to inhibit most of the CDKs evaluated, although some less potently than others.91,92 Although flavopiridol initially failed to demonstrate significant efficacy when used as a single agent,89 recent pharmacokinetic data demonstrated activity in some patients, particularly those with chronic lymphocytic leukemia (CLL), when an alternative dosing schedule that enhanced the area under the curve was employed.96 The inhibitors discussed previously tend to affect a restricted set of kinases, although other drugs exhibit much more broadly targeted inhibition. Sunitinib (Sutent) is a small molecule inhibitor of several RTKs, including VEGFR1, VEGFR2, PDGFRα/β, KIT, FLT3, receptor of colony-stimulating factor 1 (CSFR1), and rearranged during transfection receptor (RET).97 The multitargeted nature of this inhibitor may be responsible for its observed activity in several types of cancer, including GIST, renal cell carcinoma, thyroid carcinoma, and insulinoma, among others.97 Although such agents often have significant clinical activity, they are typically associated with a broader range of toxicities that may limit their use. There are now two small molecule inhibitors approved for use in dogs in veterinary medicine. Toceranib (Palladia) is a multitargeted inhibitor closely related to sunitinib that exhibits a similar target profile, including VEGFR, PDGFR, KIT, FLT3, and CSF1R. Toceranib has demonstrated activity against MCT, as well as sarcomas and carcinomas. In the original phase I study, 28% of dogs experienced objective responses to treatment, with an additional 26% experiencing stable disease for an overall biologic activity of 54%.98 A pivotal study of toceranib was subsequently conducted in dogs with recurrent or metastatic intermediate or high grade MCT resulting in an objective response rate of 42.8% (21 complete responses, 42 partial responses), with an additional 16 dogs experiencing stable disease for an overall biologic activity of 60%.99 Dogs whose MCT harbored activating mutations in KIT were roughly twice as likely to respond to toceranib than those without mutation (69% versus 37%). Following approval of toceranib in 2009, it has been used to treat a number of different solid tumors.100 Preliminary observations of biologic activity were reported in dogs with anal sac adenocarcinoma, thyroid carcinoma, head and neck carcinoma, nasal carcinoma, and OSA. Several studies are ongoing to more clearly define the role of toceranib in the treatment of canine and feline cancer. Masitinib (Kinavet) is a small molecule inhibitor of KIT, PDGFRα/β, and Lyn. In dogs with MCTs, masitinib significantly improved time to progression compared to placebo, and outcome was improved in dogs with MCT harboring KIT mutations.101 Subsequent follow-up of patients treated with long-term masitinib identified an increased number of patients with long-term disease control compared to those treated with placebo (40% versus 15% alive at 2 years).102 Finally, small studies have evaluated the efficacy of imatinib for the treatment of canine and feline MCT.103–105 Imatinib was well tolerated, and objective antitumor responses were observed in dogs with both mutant and wild-type KIT. Responses have also been observed in cats with MCT.106,107 1. Manning, G, Whyte, DB, Martinez, R, et al. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. 2. Lemmon, MA, Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117–1134. 3. Madhusudan, S, Ganesan, TS. Tyrosine kinase inhibitors in cancer therapy. Clin Biochem. 2004;37:618–635. 4. Zwick, E, Bange, J, Ullrich, A. Receptor tyrosine kinases as targets for anticancer drugs. Trends Mol Med. 2002;8:17–23. 5. Barreca, A, Lasorsa, E, Riera, L, et al. Anaplastic lymphoma kinase in human cancer. J Mol Endocrinol. 2011;47:R11–R23. 6. Fletcher, JA. Role of KIT and platelet-derived growth factor receptors as oncoproteins. Semin Oncol. 2004;31:4–11. 7. Laskin, JJ, Sandler, AB. Epidermal growth factor receptor: a promising target in solid tumours. Cancer Treat Rev. 2004;30:1–17. 8. Ma, PC, Jagadeeswaran, R, Jagadeesh, S, et al. Functional expression and mutations of c-Met and its therapeutic inhibition with SU11274 and small interfering RNA in non-small cell lung cancer. Cancer Res. 2005;65:1479–1488. 9. Ma, PC, Maulik, G, Christensen, J, et al. c-Met: structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 2003;22:309–325. 10. Thurston, G, Gale, NW. Vascular endothelial growth factor and other signaling pathways in developmental and pathologic angiogenesis. Int J Hematol. 2004;80:7–20. 11. Eskens, FA. Angiogenesis inhibitors in clinical development; where are we now and where are we going? Br J. Cancer. 2004;90:1–7. 12. Cherrington, JM, Strawn, LM, Shawver, LK. New paradigms for the treatment of cancer: the role of anti-angiogenesis agents. Adv Cancer Res. 2000;79:1–38. 13. McCarty, MF, Liu, W, Fan, F, et al. Promises and pitfalls of anti-angiogenic therapy in clinical trials. Trends Mol Med. 2003;9:53–58. 14. Thurston, G. Role of Angiopoietins and Tie receptor tyrosine kinases in angiogenesis and lymphangiogenesis. Cell Tissue Res. 2003;314:61–68. 15. Blume-Jensen, P, Hunter, T. Oncogenic kinase signalling. Nature. 2001;411:355–365. 16. Johnson, GL, Lapadat, R. Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 2002;298:1911–1912. 17. Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat Rev Cancer. 2003;3:11–22. 18. Davies, H, Bignell, GR, Cox, C, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. 19. Kumar, R, Angelini, S, Snellman, E, et al. BRAF mutations are common somatic events in melanocytic nevi. J Invest Dermatol. 2004;122:342–348. 20. Mercer, KE, Pritchard, CA. Raf proteins and cancer: B-Raf is identified as a mutational target. Biochim Biophys Acta. 2003;1653:25–40. 21. Fresno Vara, JA, Casado, E, de Castro, J, et al. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30:193–204. 22. Franke, TF, Hornik, CP, Segev, L, et al. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22:8983–8998. 23. Mitsiades, CS, Mitsiades, N, Koutsilieris, M. The Akt pathway: molecular targets for anti-cancer drug development. Curr Cancer Drug Targets. 2004;4:235–256. 24. Markman, B, Atzori, F, Perez-Garcia, J, et al. Status of PI3K inhibition and biomarker development in cancer therapeutics. Ann Oncol. 2010;21:683–691. 25. Simpson, L, Parsons, R. PTEN: Life as a tumor suppressor. Exp Cell Res. 2001;264:29–41. 26. Weng, LP, Smith, WM, Dahia, PL, et al. PTEN suppresses breast cancer cell growth by phosphatase activity-dependent G1 arrest followed by cell death. Cancer Res. 1999;59:5808–5814. 27. Kanae, Y, Endoh, D, Yokota, H, et al. Expression of the PTEN tumor suppressor gene in malignant mammary gland tumors of dogs. Am J Vet Res. 2006;67:127–133. 28. Koenig, A, Bianco, SR, Fosmire, S, et al. Expression and significance of p53, rb, p21/waf-1, p16/ink-4a, and PTEN tumor suppressors in canine melanoma. Vet Pathol. 2002;39:458–472. 29. Levine, RA, Forest, T, Smith, C. Tumor suppressor PTEN is mutated in canine osteosarcoma cell lines and tumors. Vet Pathol. 2002;39:372–378. 30. Swanton, C. Cell-cycle targeted therapies. Lancet Oncol. 2004;5:27–36. 31. Ortega, S, Malumbres, M, Barbacid, M. Cyclin D-dependent kinases, INK4 inhibitors and cancer. Biochim Biophys Acta. 2002;1602:73–87. 32. Malumbres, M, Barbacid, M. To cycle or not to cycle: a critical decision in cancer. Nat Rev Cancer. 2001;1:222–231. 33. Wellbrock, C, Ogilvie, L, Hedley, D, et al. V599EB-RAF is an oncogene in melanocytes. Cancer Res. 2004;64:2338–2342. 34. Pollock, PM, Meltzer, PS. A genome-based strategy uncovers frequent BRAF mutations in melanoma. Cancer Cell. 2002;2:5–7. 35. Wan, PT, Garnett, MJ, Roe, SM, et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell. 2004;116:855–867. 36. Dhillon, AS, Kolch, W. Oncogenic B-Raf mutations: crystal clear at last. Cancer Cell. 2004;5:303–304. 37. Brose, MS, Volpe, P, Feldman, M, et al. BRAF and RAS mutations in human lung cancer and melanoma. Cancer Res. 2002;62:6997–7000. 38. Malumbres, M, Barbacid, M. RAS oncogenes: the first 30 years. Nat Rev Cancer. 2003;3:459–465. 39. Galli, SJ, Zsebo, KM, Geissler, EN. The kit ligand, stem cell factor. Adv Immunol. 1994;55:1–95. 40. Downing, S, Chien, MB, Kass, PH, et al. Prevalence and importance of internal tandem duplications in exons 11 and 12 of c-kit in mast cell tumors of dogs. Am J Vet Res. 2002;63:1718–1723. 41. London, CA, Galli, SJ, Yuuki, T, et al. Spontaneous canine mast cell tumors express tandem duplications in the proto-oncogene c-kit. Exp Hematol. 1999;27:689–697. 42. Zemke, D, Yamini, B, Yuzbasiyan-Gurkan, V. Mutations in the juxtamembrane domain of c-KIT are associated with higher grade mast cell tumors in dogs. Vet Pathol. 2002;39:529–535. 43. Demetri, GD. Targeting the molecular pathophysiology of gastrointestinal stromal tumors with imatinib. Mechanisms, successes, and challenges to rational drug development. Hematol Oncol Clin North Am. 2002;16:1115–1124. 44. Demetri, GD. Differential properties of current tyrosine kinase inhibitors in gastrointestinal stromal tumors. Semin Oncol. 2011;38(Suppl 1):S10–S19. 45. Frost, D, Lasota, J, Miettinen, M. Gastrointestinal stromal tumors and leiomyomas in the dog: a histopathologic, immunohistochemical, and molecular genetic study of 50 cases. Vet Pathol. 2003;40:42–54. 46. Kondo, M, Horibe, K, Takahashi, Y, et al. Prognostic value of internal tandem duplication of the FLT3 gene in childhood acute myelogenous leukemia. Med Pediatr Oncol. 1999;33:525–529. 47. Nakoa, M, Yokota, S, Iwai, T, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911–1918. 48. Yokota, S, Kiyoi, H, Nakao, M, et al. Internal tandem duplication of the FLT3 gene is preferentially seen in acute myeloid leukemia and myelodysplastic syndrome among various hematological malignancies. A study on a large series of patients and cell lines. Leukemia. 1997;11:1605–1609. 49. Iwai, T, Yokota, S, Nakao, M, et al. Internal tandem duplication of the FLT3 gene and clinical evaluation in childhood acute myeloid leukemia. The Children’s Cancer and Leukemia Study Group, Japan. Leukemia. 1999;13:38–43. 50. Pao, W, Chmielecki, J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10:760–774. 51. Wen, J, Fu, J, Zhang, W, et al. Genetic and epigenetic changes in lung carcinoma and their clinical implications. Mod Pathol. 2011;24:932–943. 52. Paik, S, Hazan, R, Fisher, ER, et al. Pathologic findings from the National Surgical Adjuvant Breast and Bowel Project: prognostic significance of erbB-2 protein overexpression in primary breast cancer. J Clin Oncol. 1990;8:103–112. 53. Slamon, DJ, Clark, GM, Wong, SG, et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–182. 54. Libermann, TA, Nusbaum, HR, Razon, N, et al. Amplification, enhanced expression and possible rearrangement of EGF receptor gene in primary human brain tumours of glial origin. Nature. 1985;313:144–147. 55. Libermann, TA, Nusbaum, HR, Razon, N, et al. Amplification and overexpression of the EGF receptor gene in primary human glioblastomas. J Cell Sci Suppl. 1985;3:161–172. 56. Golub, TR, Barker, GF, Lovett, M, et al. Fusion of PDGF receptor beta to a novel ets-like gene, tel, in chronic myelomonocytic leukemia with t(5;12) chromosomal translocation. Cell. 1994;77:307–316. 57. Gotlib, J, Cools, J, Malone, JM, 3rd., et al. The FIP1L1-PDGFRalpha fusion tyrosine kinase in hypereosinophilic syndrome and chronic eosinophilic leukemia: implications for diagnosis, classification, and management. Blood. 2004;103:2879–2891. 58. Melo, JV, Hughes, TP, Apperley, JF. Chronic myeloid leukemia. Hematology (Am Soc Hematol Educ Program). 2003:132–152. [2003]. 59. Van Etten, RA. Mechanisms of transformation by the BCR-ABL oncogene: new perspectives in the post-imatinib era. Leuk Res. 2004;28(Suppl 1):S21–S28. [2004]. 60. Medves, S, Demoulin, JB. Tyrosine kinase gene fusions in cancer: Translating mechanisms into targeted therapies. J Cell Mol Med. 2012;16(2):237–248. 61. Sciacca, L, Costantino, A, Pandini, G, et al. Insulin receptor activation by IGF-II in breast cancers: evidence for a new autocrine/paracrine mechanism. Oncogene. 1999;18:2471–2479. 62. Ekstrand, AJ, James, CD, Cavenee, WK, et al. Genes for epidermal growth factor receptor, transforming growth factor alpha, and epidermal growth factor and their expression in human gliomas in vivo. Cancer Res. 1991;51:2164–2172. 63. Graeven, U, Fiedler, W, Karpinski, S, et al. Melanoma-associated expression of vascular endothelial growth factor and its receptors FLT-1 and KDR. J Cancer Res Clin Oncol. 1999;125:621–629. 64. Fosmire, SP, Dickerson, EB, Scott, AM, et al. Canine malignant hemangiosarcoma as a model of primitive angiogenic endothelium. Lab Invest. 2004;84:562–572. 65. MacEwen, EG, Kutzke, J, Carew, J, et al. c-Met tyrosine kinase receptor expression and function in human and canine osteosarcoma cells. Clin Exp Metastasis. 2003;20:421–430. 66. Ferracini, R, Angelini, P, Cagliero, E, et al. MET oncogene aberrant expression in canine osteosarcoma. J Orthop Res. 2000;18:253–256. 67. Harris, M. Monoclonal antibodies as therapeutic agents for cancer. Lancet Oncol. 2004;5:292–302. 68. Vogel, CL, Cobleigh, MA, Tripathy, D, et al. First-line Herceptin monotherapy in metastatic breast cancer. Oncology. 2001;61(Suppl 2):37–42. 69. Slamon, DJ, Leyland-Jones, B, Shak, S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;2001(344):783–792. 70. Arteaga, CL, Sliwkowski, MX, Osborne, CK, et al. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2011;9(1):16–32. 71. Mukai, H. Treatment strategy for HER2-positive breast cancer. Int J Clin Oncol. 2010;15:335–340. 72. Cabanillas, F. Front-line management of diffuse large B cell lymphoma. Curr Opin Oncol. 2010;22:642–645. 73. Vidal, L, Gafter-Gvili, A, Salles, G, et al. Rituximab maintenance for the treatment of patients with follicular lymphoma: an updated systematic review and meta-analysis of randomized trials. J Natl Cancer Inst. 2001;103:1799–1806. 74. Brand, TM, Iida, M, Wheeler, DL. Molecular mechanisms of resistance to the EGFR monoclonal antibody cetuximab. Cancer Biol Ther. 2011;11:777–792. 75. Zhang, J, Yang, PL, Gray, NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28–39. 76. de Kogel, CE, Schellens, JH. Imatinib. Oncologist. 2007;12:1390–1394. 77. Mauro, MJ, Druker, BJ. STI571: targeting BCR-ABL as therapy for CML. Oncologist. 2001;6:233–238. 78. Kantarjian, H, Sawyers, C, Hochhaus, A, et al. Hematologic and cytogenetic responses to imatinib mesylate in chronic myelogenous leukemia. N Engl J Med. 2002;346:645–652. 79. Beham-Schmid, C, Apfelbeck, U, Sill, H, et al. Treatment of chronic myelogenous leukemia with the tyrosine kinase inhibitor STI571 results in marked regression of bone marrow fibrosis. Blood. 2002;99:381–383. 80. Druker, BJ, Talpaz, M, Resta, DJ, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. 81. Druker, BJ, Sawyers, CL, Kantarjian, H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. N Engl J Med. 2001;344:1038–1042. 82. Sawyers, CL. Rational therapeutic intervention in cancer: Kinases as drug targets. Curr Opin Genet Dev. 2002;12:111–115. 83. Weisberg, E, Griffin, JD. Resistance to imatinib (Glivec): update on clinical mechanisms. Drug Resist Updat. 2003;6:231–238. 84. Nardi, V, Azam, M, Daley, GQ. Mechanisms and implications of imatinib resistance mutations in BCR-ABL. Curr Opin Hematol. 2004;11:35–43. 85. Duffaud, F, Blay, JY. Gastrointestinal stromal tumors: Biology and treatment. Oncology. 2003;65:187–197. 86. Heinrich, MC, Rubin, BP, Longley, BJ, et al. Biology and genetic aspects of gastrointestinal stromal tumors: KIT activation and cytogenetic alterations. Hum Pathol. 2002;33:484–495. 87. Miettinen, M, Sarlomo-Rikala, M, Lasota, J. Gastrointestinal stromal tumors: recent advances in understanding of their biology. Hum Pathol. 1999;30:1213–1220. 88. Miettinen, M, Sarlomo-Rikala, M, Lasota, J. Gastrointestinal stromal tumours. Ann Chir Gynaecol. 1998;87:278–281. 89. Heinrich, MC, Corless, CL, Duensing, A, et al. PDGFRA activating mutations in gastrointestinal stromal tumors. Science. 2003;299:708–710. 90. Peled, N, Yoshida, K, Wynes, MW, et al. Predictive and prognostic markers for epidermal growth factor receptor inhibitor therapy in non-small cell lung cancer. Ther Adv Med Oncol. 2009;1:137–144. 91. Bang, YJ. The potential for crizotinib in non-small cell lung cancer: A perspective review. Ther Adv Med Oncol. 2011;3:279–291. 92. Shaw, AT, Yeap, BY, Solomon, BJ, et al. Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis. Lancet Oncol. 2011;12:1004–1012. 93. Chapman, PB, Hauschild, A, Robert, C, et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. 94. Markman, B, Dienstmann, R, Tabernero, J. Targeting the PI3K/Akt/mTOR pathway–beyond rapalogs. Oncotarget. 2010;1:530–543. 95. Vilar, E, Perez-Garcia, J, Tabernero, J. Pushing the envelope in the mTOR pathway: the second generation of inhibitors. Mol Cancer Ther. 2011;10:395–403. 96. Christian, BA, Grever, MR, Byrd, JC, et al. Flavopiridol in the treatment of chronic lymphocytic leukemia. Curr Opin Oncol. 2007;19:573–578. 97. Papaetis, GS, Syrigos, KN. Sunitinib: a multitargeted receptor tyrosine kinase inhibitor in the era of molecular cancer therapies. BioDrugs. 2009;23:377–389. 98. London, CA, Hannah, AL, Zadovoskaya, R, et al. Phase I dose-escalating study of SU11654, a small molecule receptor tyrosine kinase inhibitor, in dogs with spontaneous malignancies. Clin Cancer Res. 2003;9:2755–2768. 99. London, CA, Malpas, PB, Wood-Follis, SL, et al. Multi-center, placebo-controlled, double-blind, randomized study of oral toceranib phosphate (SU11654), a receptor tyrosine kinase inhibitor, for the treatment of dogs with recurrent (either local or distant) mast cell tumor following surgical excision. Clin Cancer Res. 2009;15:3856–3865. 100. London, C, Mathie, T, Stingle, N, et al. Preliminary evidence for biologic activity of toceranib phosphate (Palladia) in solid tumours. Vet Comp Oncol. 2011. [Epub ahead of print June 1]. 101. Hahn, KA, Ogilvie, G, Rusk, T, et al. Masitinib is safe and effective for the treatment of canine mast cell tumors. J Vet Intern Med. 2008;22:1301–1309. 102. Hahn, KA, Legendre, AM, Shaw, NG, et al. Evaluation of 12- and 24-month survival rates after treatment with masitinib in dogs with nonresectable mast cell tumors. Am J Vet Res. 2010;71:1354–1361. 103. Isotani, M, Ishida, N, Tominaga, M, et al. Effect of tyrosine kinase inhibition by imatinib mesylate on mast cell tumors in dogs. J Vet Intern Med. 2008;22(4):985–988. 104. Marconato, L, Bettini, G, Giacoboni, C, et al. Clinicopathological features and outcome for dogs with mast cell tumors and bone marrow involvement. J Vet Intern Med. 2008;22(4):1001–1007. 105. Yamada, O, Kobayashi, M, Sugisaki, O, et al. Imatinib elicited a favorable response in a dog with a mast cell tumor carrying a c-kit c.1523A>T mutation via suppression of constitutive KIT activation. Vet Immunol Immunopathol. 2011;142:101–106. 106. Isotani, M, Tamura, K, Yagihara, H, et al. Identification of a c-kit exon 8 internal tandem duplication in a feline mast cell tumor case and its favorable response to the tyrosine kinase inhibitor imatinib mesylate. Vet Immunol Immunopathol. 2006;114:168–172. 107. Isotani, M, Yamada, O, Lachowicz, JL, et al. Mutations in the fifth immunoglobulin-like domain of kit are common and potentially sensitive to imatinib mesylate in feline mast cell tumours. Br J Haematol. 2009;148:144–153.
Molecular/Targeted Therapy of Cancer
 Section A
Section A
Gene Therapy for Cancer
Efficient Gene Delivery: The Major Hurdle to Clinical Benefits
Viral Vectors
Nonviral Gene Delivery
Targeted Gene Delivery
Gene Therapy Strategies for Cancer
Rescue of the Cancer Cell Through Gene Replacement Technologies
Destruction of Cancer Cells Through Delivery of “Suicide Genes”
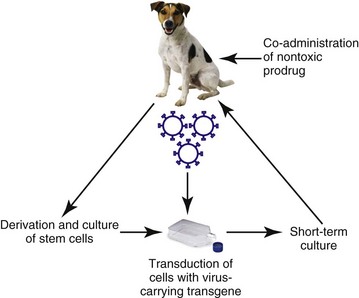
Utilizing Stem Cells to Deliver “Suicide Genes”
Gene-Directed Immunotherapy
Delivery of Chemoprotective Genes
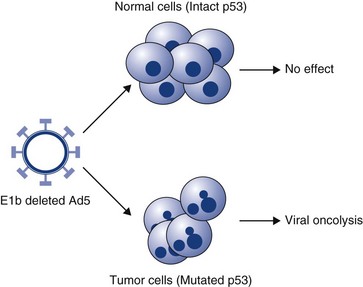
The Use of Replication-Competent Viral Vectors
Safety Considerations in Gene Therapy
References
 Section B
Section B
Signal Transduction and Cancer
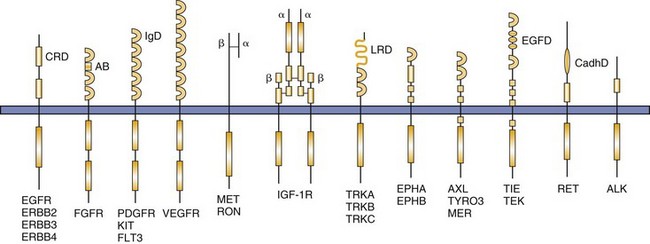
Protein Kinases and Normal Cells
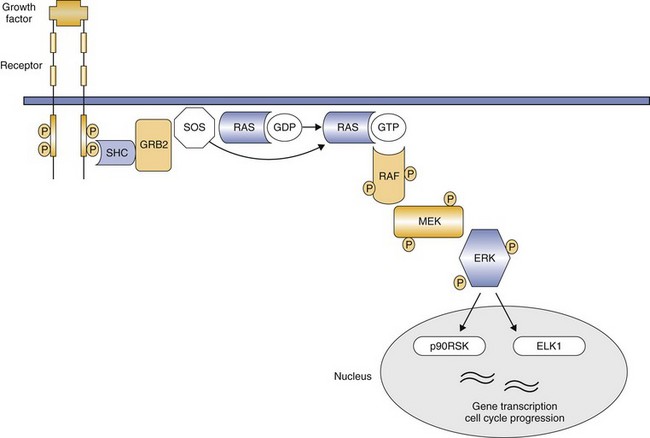
Protein Kinases and Cancer Cells
Tyrosine Kinase
Cancer Association
EGFR family
Breast, ovary, lung, stomach, colon, glioblastoma
Insulin receptor family
Sarcomas, cervix, kidney
PDGFR family
Glioblastoma, ovary, CMML, GIST
KIT
AML, GIST, seminoma, MCT, melanoma
Flt3
AML
VEGFR family
Angiogenesis, Kaposi’s sarcoma, hemangiosarcoma, melanoma
FGFR family
AML, lymphoma, breast, prostate, multiple myeloma, TCC
NGFR family
Thyroid cancer, neuroblastoma, fibrosarcoma, AML
Met/Ron
Thyroid cancer, osteosarcoma, rhabdomyosarcoma, liver, kidney, colon
EPHR family
Melanoma, stomach, colon, breast, esophagus
AXL
AML
Tie family
Angiogenesis, stomach, hemangioblastoma
RET family
Thyroid cancer, multiple endocrine neoplasia
ALK
Non-Hodgkin’s lymphoma, lung
Inhibition of Kinases
Kinase Inhibitors in Veterinary Medicine
References
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Molecular/Targeted Therapy of Cancer