CHAPTER 58 Metabolic Encephalopathy
Organic Acidurias
The organic acidurias are a heterogeneous group of diseases characterized by excretion of abnormal organic acids in the urine.1–3 Organic acids can be either excessive amounts of a metabolite found normally in the urine, or one that would not be expected in the urine of a normal cat. The abnormal organic acid in the blood and/or the underlying metabolic disease that produced it can lead to altered brain function and signs of metabolic encephalopathy. In this chapter we will review the role of organic acids in metabolism, and the pathogenesis, recognition, and treatment of organic acidurias.
ORGANIC ACIDS
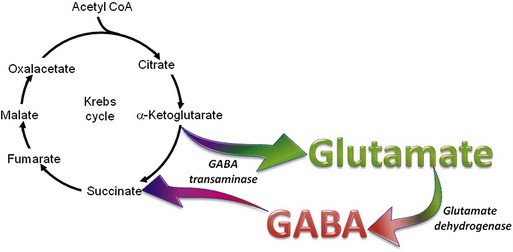
The organic acids are vital compounds in intermediate metabolism—the intermediate steps in conversion of nutrients into cellular components and energy (Figure 58-1). At the center of intermediate metabolism is the Krebs cycle. Fats, carbohydrates, and proteins in the diet are broken down into organic acids that enter the Krebs cycle at various points and sometimes are conjugated with coenzyme A (CoA). Hydrogen molecules generated from the metabolism of these acids in the mitochondria are fed into the respiratory chain to yield adenosine triphosphate (ATP) needed to supply the energy needs of cells. In addition to their role in energy production, the organic acids of intermediate metabolism can be crucial steps in the synthesis of cellular components such as neurotransmitters. For example, gamma aminobutyric acid (GABA) and glutamate, respectively the most important inhibitory and excitatory neurotransmitters in the brain, are synthesized from α-ketoglutarate in the “GABA shunt” (Figure 58-2).
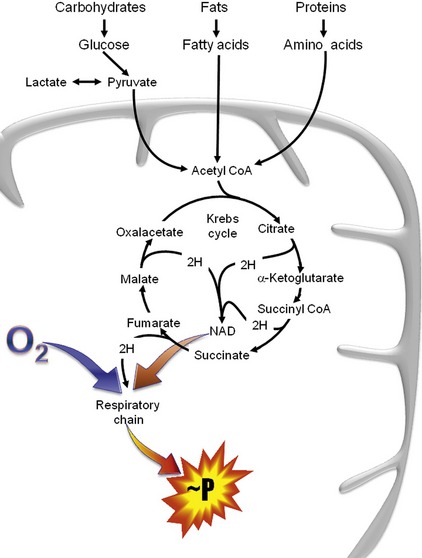
Several of the B-vitamins are significant cofactors in intermediate metabolism. Thiamin (vitamin B1) is a cofactor for several dehydrogenase reactions, most importantly the conversion of α-ketoglutarate to succinyl CoA in the Krebs cycle.4 Neonates are particularly sensitive to biotin (vitamin B7) deficiency, which is essential to activate a number of carboxylases necessary for normal gluconeogenesis, fatty acid synthesis, and amino acid catabolism.5 Cobalamin (vitamin B12) is necessary for conversion of methylmalonyl CoA to succinyl CoA, the entry to the Krebs cycle for cholesterol and some fatty acids and amino acids (see below).4
DIAGNOSING ORGANIC ACID DISORDERS
Because they are small molecules, the organic acids are excreted readily in the urine. This is in contrast to other errors of metabolism such as the lysosomal storage diseases in which the byproduct is a large macromolecule that accumulates within the cell. Specialized laboratories can detect the abnormal organic acids in urine by gas chromatography/mass spectroscopy. The pattern of organic acids found indicates the nature of the disturbance and sometimes can indicate a specific enzyme or vitamin deficiency as the source of the disorder. Although routine clinical pathology tests typically do not detect organic acids, they may contain clues that should lead to a urine organic acid assay (Box 58-1). An unexplained metabolic acidosis should raise suspicion of an organic acid disorder. The unknown acid could produce a high anion gap or low bicarbonate or total CO2 on a serum biochemistry panel, and a blood gas analysis would reveal a metabolic acidosis with respiratory compensation. Interference with energy production or utilization could produce hypoglycemia or ketosis, which would be detected on urinalysis. If the urea cycle is affected, increased blood ammonia could result without other signs of liver disease.
D-LACTIC ACIDOSIS WITH GASTROINTESTINAL DISEASE
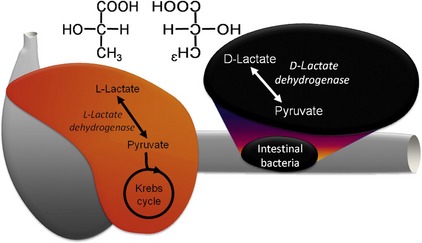
We have seen two cats with encephalopathy associated with gastrointestinal disease and profound metabolic acidosis. Serum lactate levels, as measured by routine bench-top assay, were normal, and yet very high levels of lactate were detected in the urine.6 This presented a quandary about the discrepancy between the serum and urine tests and about the source of the lactate elevation. Neither cat was in shock or otherwise hypoxemic, and both cats had been clinically normal for years making an inborn error of metabolism unlikely.
Lactic acid can exist as one of two stereoisomers, L-lactate and D-lactate (Figure 58-3), and enzymes can only metabolize one isoform or the other. In mammals L-lactate dehydrogenase in the cytoplasm converts L-lactate efficiently to pyruvate, which enters the mitochondria to fuel the Krebs cycle. When there is insufficient oxygen for aerobic metabolism such as hypoxemia, shock, or extreme muscle activity, this reaction can be reversed resulting in the lactic acidosis. The traditional concept that neurons rely exclusively on glucose for energy has been challenged by the “lactate shuttle” hypothesis.7 In this paradigm, L-lactate provides up to 75 per cent of the energy requirements of neurons. The excitatory neurotransmitter glutamate is taken up from the synapse by astrocytes to be recycled, but it also stimulates conversion of pyruvate to L-lactate within the astrocyte. This lactate is shuttled to neurons through specific lactate transporters, where it is metabolized readily to pyruvate and then through the Krebs cycle to produce the energy needed to maintain the resting membrane potential. Therefore this system links energy supply with demand produced by excitation.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


