CHAPTER 11 Laboratory Diagnosis of Viral Infections
The ability to diagnose viral infections is important for equine clinicians. Accurate and timely laboratory confirmation of a viral infection allows more effective supportive clinical management, earlier detection of complications, more accurate prognosis, more effective isolation protocols, evaluation of vaccination programs, detection of vaccination failures, and probably in the future, selection of appropriate antiviral drugs.
VIRUS ISOLATION
The isolation of a virus in cell culture is the original virus detection assay.1 The basis for this method is to inoculate a clinical specimen onto living cells in which the virus can propagate, then detect and identify any viruses that are isolated. Living cell cultures are first established in plastic wells or Petri dishes. Clinical samples are homogenized in cell culture growth media, centrifuged or filtered to remove larger debris and bacteria, treated with antibiotics to remove contaminating bacteria that may infect and destroy the cell cultures, and then inoculated onto the cells. Viral growth in the cells is detected by characteristic changes in the infected cells, called the cytopathic effect (CPE), caused by the virus (Fig. 11-1). When viral CPE is suspected in a cell culture, the presence of a virus as well as its identity is confirmed by additional tests, such as fluorescent-labeled antiviral antibodies, PCR for viral nucleic acids, or electron microscopy. Occasionally, clinical samples are inoculated into laboratory animals for isolation of viruses, for example, into the brain of suckling mice for isolation of alphaviruses (e.g., eastern and western equine encephalitis viruses)2 or into embryonated eggs for isolation of equine influenza virus.3 Depending on the virus or virus strain and the specific cells being used for isolation, viral growth may not cause any visible changes and is called noncytopathic. For these virus/cell combinations, screening for the presence of virus must be done in all inoculated cell cultures by some other method, such as immunofluorescence for viral antigens or PCR for viral nucleic acids.
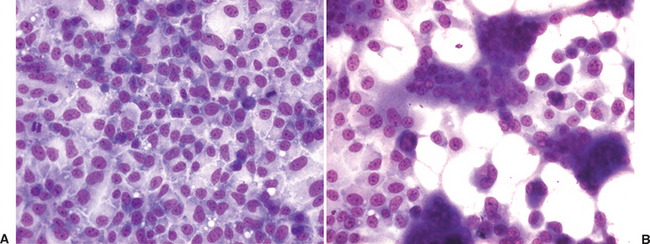
One of the primary advantages of virus isolation in cell culture is the potential for very good sensitivity. Because inoculated cell cultures are generally passaged serially two or three times before being called “negative,” there is usually significant amplification of any virus present in the sample. However, virus isolation requires that viable virus be present in the sample, and viability may be adversely affected by sample collection and storage, timing of sample collection, postmortem interval, and sample transport. This is particularly true for fragile viruses such as herpesviruses and other enveloped viruses. Therefore, good sample collection and transport are critical for successful virus isolation. Sensitivity is also affected by the type of cells used for isolation. Different cell types vary in their ability to propagate different viruses and thus in their sensitivity of detection.3 Therefore, most laboratories will use specific types of cultured cells or animal inoculation systems for specific viruses and thus require some guidance from the clinician about the suspected viruses.
Another advantage of cell culture is that it obtains an isolate of viable virus for evaluation of biologic properties, such as virulence, antigenic variation, and possibly, susceptibility to antiviral drugs.1 Fortunately, with most of the common equine viral diseases, this additional information is not essential for the effective clinical management of individual cases. However, for viruses such as equine influenza or African horse sickness, the ability to type a virus or detect changes in antigenicity may explain vaccine failures or lead to changes in vaccine type.4,5 Usually, obtaining isolates for this purpose is indicated only when there is a significant and unexplained change in the epidemiology of a particular disease. Virus isolation is also a relatively nonselective method of virus detection, which is important for the identification of new viruses. However, the search for a new virus is usually based first on unexplained viral pathology or clinical features that suggest a viral etiology. Whether the disease is caused by a change in the epidemiology of a recognized viral disease, or the appearance of a new virus, the associated investigations generally become research projects and have different goals (and sources of funding) than the routine detection of recognized equine viral pathogens.
POLYMERASE CHAIN REACTION
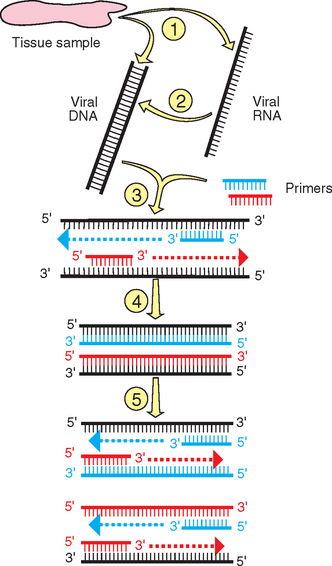
Nucleic acid amplification by PCR is now one of the most common assays used to detect the presence of a virus directly in clinical samples. PCR assays have been developed for a variety of equine viral pathogens from a wide array of sample types3,4,6–16 (Fig. 11-2).
Amplification Technique
Total deoxyribonucleic acid (DNA) or ribonucleic acid (RNA), including viral DNA or RNA if present, is extracted and purified from the sample. For RNA viruses, isolated RNA is converted to complementary DNA (cDNA) in a separate reaction by the enzyme reverse transcriptase (RT); therefore, PCR for RNA targets is referred to as RT-PCR. A heat-stable DNA polymerase is then used in a series of cyclic reactions to amplify exponentially any viral DNA or cDNA sequences. The reaction is initiated and driven by two oligonucleotide primers (usually about 15-25 nucleotides in length) that are synthetically produced and complementary to a desired target sequence in the viral genome. This target sequence is selected to be unique for the virus, preventing the primers from binding to and initiating amplification of nonviral sequences in the genome of the host or other pathogens. When the primers bind to the target DNA, they allow the DNA polymerase to initiate copying of the target sequence from the 3′ end of the primer. The primers are also designed to bind to opposite strands of the double-stranded DNA target and thus initiate reactions toward each other. The heat stability of the DNA polymerase is required to prevent inactivation at the high temperatures (about 90° C) used to denature the double-stranded target for primer binding. High temperatures (usually about 55°-65° C) during the annealing part of the reaction are also used to maintain the fidelity of primer binding, because at these temperatures, binding will occur only if there is a perfect complementary match between the primer and target sequences. The other parameter important for primer selection is that they bind close enough on the target genome so that the copying reactions will efficiently copy up to the binding sites for the other primer on the opposite DNA strand. However, the amplicons also need to remain large enough to be detected. The optimal size for a PCR amplicon is about 150 to 800 base pairs. Because amplification is exponential, the final result of the PCR assay is a very large number of amplified DNA fragments of a specific length (i.e., the distance between the primers).
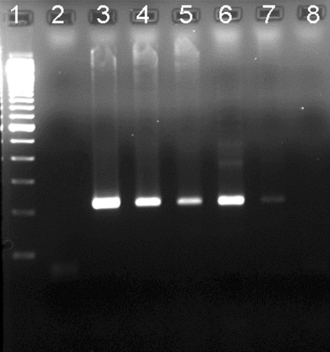
The amplicon products of PCR amplification can then be detected in several ways. The most common method is agarose gel electrophoresis, which separates DNA fragments based on size (Fig. 11-3). Positive reactions are identified by the detection of DNA bands of the correct size or that hybridize to sequence-specific probes.
Design Variations
Several variants of the PCR assay are designed to decrease the time necessary to perform the assay, minimize manipulations of reactions, and improve sensitivity. Decreased manipulations not only save time and labor, but more importantly, reduce the amount of amplicon contamination that inevitably occurs when opening and closing finished reactions and transferring reaction products to new tubes or analytical gels. Contaminant amplicons are notorious for their ability to get into reagents and other samples, leading to false-positive results. To avoid reaction manipulations, some assays for RNA viruses now use one-step protocols or polymerase enzymes with both RT and DNA polymerase activities, eliminating the need for separate reactions.16–18 A common variant now is “real-time” PCR, in which amplification of either the target DNA or the cDNA is detected by the formation of a fluorescent signal in the tube, and the level of fluorescence is measured by the PCR machine during each cycle.6,16,19 This not only eliminates the need for opening amplified reactions for gel analysis, but also allows positive and negative tests to be determined as the reaction proceeds, thus in “real time.”
The sensitivity of PCR assays can be maximized by “nested” PCR.3,7 This modification uses two sets of virus-specific primers. The first set of primers is used for an initial round of amplification as previously described. Some of this reaction product is transferred to a second PCR assay with a second set of primers that will bind to the first amplicon. Because there are two rounds of amplification, this modification allows detection of very low levels of virus. However, because of the extreme sensitivity of nested PCR, this assay is prone to false-positive results from minute levels of amplicon contamination that are very difficult to control in most laboratories. Moreover, good-quality samples from active viral infections do not usually require the sensitivity of nested PCR for detection. Thus, most laboratories avoid nested PCR assays, if possible.
Other recent variants of PCR used in human laboratories include the ligase chain reaction, strand-displacement amplification, transcription-mediated amplification, and microarray analysis.1,19 However, these assays are not yet in widespread use for the detection of equine viruses and thus are not considered further here.
Advantages and Benefits
PCR assays are generally quite sensitive because of logarithmic amplification of the target and are often comparable in sensitivity to virus isolation. However, this is not automatically true, and the performance of PCR must be assessed for each pathogen and disease. PCR is especially useful for viruses that are either slow, difficult, or unable to be propagated in cell culture, such as many of the gamma herpesviruses (e.g., equine herpesvirus types 2 and 5), papillomavirus, influenza virus, and rotavirus. PCR is also very useful to detect viruses that are fragile and prone to loss of viability during sampling or transport, such as many enveloped viruses. PCR for diagnosis of viruses that present significant hazards to laboratory personnel, such as West Nile and eastern equine encephalitis viruses, is preferable to cell culture techniques that require propagation of live virus. PCR can also be performed on DNA or RNA extracted from formalin-fixed, paraffin-embedded tissues.14 This option provides not only good laboratory safety, but also allows a diagnosis to be made when fresh tissue samples are not available.
Disadvantages and Limitations
PCR does not result in recovery of a viable isolate of the virus, which can be a disadvantage if biologic information about the virus is needed. However, as noted in the section on virus isolation, this information is rarely needed to manage clinical cases. Another potential problem with PCR is the detection of viruses that are not clinically relevant. This limitation is not unique to PCR, and detection of a virus in a clinical sample by any method should not automatically be assumed to be clinically relevant. For example, many adult horses may periodically shed equine herpesvirus type 1 or 4 (EHV-1 or EHV-4) subclinically in nasal secretions, and detection of these viruses by either virus isolation or PCR may or may not be important.15,20
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


