Chapter 31 Immunomodulators or Biological Response Modifiers
Introduction and Miscellaneous Agents∗
The Immune Response
Immunomodulators, or biological response modifiers, are agents or drugs that act to regulate or modify the host’s immune response to a microbe, neoplasm, or inflammatory response. It is beyond the scope of this chapter to provide a comprehensive review of the immune system. The following is a general review only. Cytokine biology can also be reviewed elsewhere,1,2 as can interferon (IFN) biology.3
Immune Defenses
Immune defenses are composed of the innate and adaptive systems. The innate systems are nonspecific in their response and are best exemplified by barriers provided by the integument; the gastrointestinal environment (acid pH, mucosal and epithelial barriers, microbiota); the mucociliary tract of the respiratory system; and the intimate vasculature of selected organs such as the placenta, brain, and prostate. Other nonspecific defense mechanisms are exemplified fever and the antimicrobial actions of many secretions. White blood cells also represent innate immunity. These include neutrophils, eosinophils, basophils, macrophages, and dendritic cells, and unconventional T-cell subsets bearing T-cell receptors (TCR), natural killer (NK; CD1d) cells and gamma/delta (γδ) T-cells. Natural killer (NK) cells are innate (non conventional) cytotoxic T cells that work in concert with the adaptive arm of immunity, targeting compromised host cells such as tumor or virally infected cells. Released in the vicinity of a cell slated for destruction, it is stimulated by macrophage cytokines. They recognize targeted cells which containing abnormally low concentrations of major histocompatibility complex [MHC] class I antigen (“missing self”). Materials that induce apoptosis enter the target cell through the pores, thus avoiding cellular lysis, which is of benefit in the presence of virally-infected cells that might release viral materials causing host re-infection. NK cells exhibiting the FrC receptor interact with the humoral arm of adaptive immunity, causing antibody-dependent cell cytotoxicity. Gamma-delta T cells contain characteristics of both the innate and adaptive systems.
Both cell-mediated and humoral immune mechanisms are characterized by specificity toward antigenic epitopes expressed as molecular components of infectious organisms, foreign (transplanted) cells, or transformed (malignant) cells. Cytokines, including soluble growth and activation factors (Table 31-1), are a vital component of the adaptive response and are released and subsequently mediate the response of the various cell populations involved in both cell-mediated and humoral immunity.4 In addition to cytokines, a number of other molecules (e.g., adhesion or accessory molecules) are necessary as “second signals” for antigen processing, recognition, or response.4
Effectors
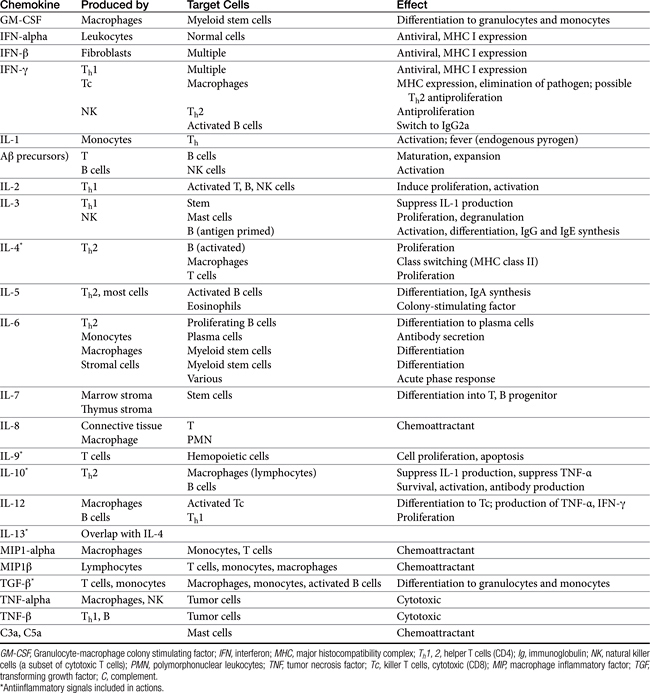
The following is a brief synopsis of the events following infection to implementation of the immune response (Figure 31-1). The antigen is exposed to an antigen-processing or presenting cell (APC), which includes dendritic cells (the principle APC), macrophages, and activated B cells.4 The antigen is identified by the APC as foreign and is subsequently phagocytized by the APC. The APC “processes” the antigen and “exhibits” it on the cell surface in a groove made by the MHC molecule. Peptides derived from endogenous cytosolic proteins synthesized within the cell complex (including those synthesized in response to viral stimuli) are expressed with class II MHC molecules on the cell surface, whereas exogenous intracellular proteins are expressed bound to a class I MHC groove for presentation.4
A single APC surface may have tens of thousands of MHC molecules, each containing a different peptide; a single animal may have more than 108 APCs. The APC migrates to the T cell area of a lymph node and presents the antigen to naïve CD4 T cells. If the APC comes into contact with a CD4 T cell with a TCR that recognizes the antigen associated with the MHC, it becomes activated. The release of adhesion molecules causes the two cells to stick together, which facilitates interaction between the APC and the CD4. Cytokines move between the two cells, and production of interleukin-1 (IL-1) by the APC and interleukin-2 (IL-2) from the activated CD4 cell itself amplifies the sequelae of CD4 activation. The activated CD4 cell differentiates, proliferates, and produces a number of cytokines (see Table 31-1), which results in recruitment of other leukocytes, initiation of B cell production of immunoglobulins (Igs), and formation of other T cell colonies, including “memory” cells (see Figure 31-1).4,5
Cellular Components
Efficient pathogen elimination depends on the adaptive immune response. Two classes of lymphocytes are responsible for adaptive immunity (see Figure 31-1). B lymphocytes must be programmed to respond to antigen exposure and, after activation to plasma cells, are responsible for the production of specific Igs (humoral response). T lymphocytes provide the primary regulation of the immune response. T cell activity begins with specific antigen recognition by a receptor on the surface of the cell.4 T cells are further subdivided into several populations of cells depending on their role in immunoregulation (see Figure 31-1). Helper cell (CD4 T cells; Th cells) receptors recognize and bind to the peptide–MHC class II complex of APC cells. In response to IL-1, CD4 cells consequently proliferate and become primed as either Th1 or Th2 CD4 cells, which modulate further responses in both the cell-mediated and humoral arms (see Figure 31-1). Migration of activated lymphocytes from lymph nodes to tissues is facilitated by adhesion molecules expressed by endothelial cells in tissues. Whether a T cell becomes Th1 or Th2 reflects the stimulating cytokine: IL-12 activates signal transducer and activator of transcription 4 (STAT-4), which regulates Th1 differentiation, whereas IL-4 activates STAT-6 and Th2 differentiation.5
The Th1 subsets of CD4 produce primarily proinflammatory cytokines such as IFN-γ (IFN-γ) and tumor necrosis factor-alpha (TNF-α) (cachexin; see Figure 31-1). As such, Th1 cells regulate signals that promote cell-mediated immunity and control intracellular pathogens. Th1 response is paramount to successful resistance to most microbial pathogens, including bacteria, intracellular protozoa, and fungal organisms. Additionally, Th1 cells mediate organ-specific autoimmunity and as such are crucial to the pathogenesis of autoimmune diseases. The cell-mediated response occurs when activated CD4 (Th1) cells attract other cells (polymorphonuclear leukocytes, eosinophils, and monocytes) to support cellular killing; the result is referred to as delayed hypersensitivity. Activated CD4 cells also yield, in response to IL-2, helper T cells that contain the glycoprotein CD8 and are responsible for the T cell–mediated cytotoxic response. Receptors of the CD8 T cells recognize peptide–MHC class I complexes (generally associated with endogenous peptides present on all nucleated cells) on APCs. Recognition of the CD8 receptor and subsequent interaction with CD4 cells result in the generation of cytolytic (CTL) or cytotoxic T cells (TC; killer T cells). Cytotoxic T cells are capable of directly (without further interaction with CD4 cells) causing lysis of cells expressing the targeted specific peptide–MHC complex.4 This includes normal but otherwise damaged (include virally infected or cells damaged by TNF-β or lymphotoxin) cells. CD8 cells release several different cytotoxins which cause pores in the target cell membrane. Cytotoxic T cells also undergo clonal expansion, resulting in prepared effector cells. In contrast to Th1 cells, Th2 cells secrete antiinflammatory cytokines (IL-4, IL-9, IL-10, IL-13) (in additional to pro-inflammatory cytokines) and support the humoral immune response, including host defense against intestinal helminths. Because of their antiinflammatory effects, Th2 cytokines can decrease autoimmune diseases associated with cell-mediated immunity; however, an imbalance toward Th2 cells have been implicated in the pathogenesis of asthma and allergy (see below).5 Dysregulation of the influence of STAT-4 and STAT-6—which have contrary effects—has been associated with immune-mediated diseases. For example, each STAT is reported to be involved in systemic lupus erythematosus: STAT-6 contributes to the development of glomerulosclerosis and antibody production, which is exacerbated in the absence of STAT-4.5
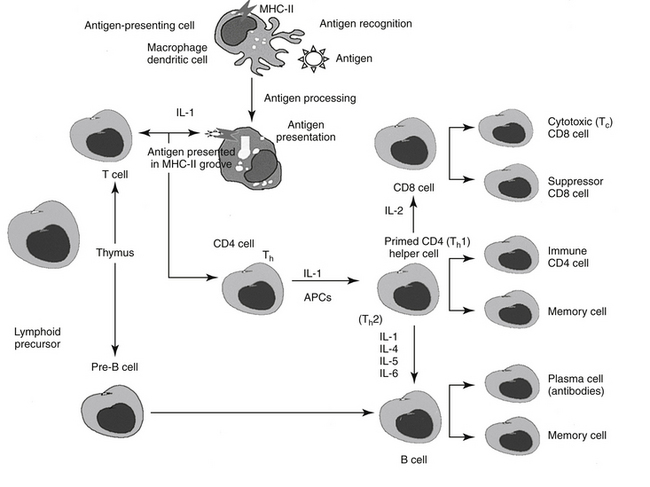
In contrast to immune-mediated diseases, in which Th1 cells play key roles, key effector cells in all chronic allergic diseases include eosinophils, basophils, and Th2 cells.6 Their involvement occurs through release of preformed, or formed in situ, granule proteins and cytokines.7 A focus on eosinophil regulation may offer targeted therapy for chronic allergic diseases. Cysteinyl leukotrienes appear to have an important role in the regulation of human eosinophil hematopoiesis, recruitment, and activation.7 Eosinophils are generated from CD34+ progenitors in the bone marrow upon stimulation by cytokines such as IL- 5. A single dose of IL-5 antibody profoundly inhibited circulating eosinophils and allergen-induced sputum eosinophils but not airway hyperreactivity in humans. In a follow-up study, eosinophil numbers in blood and bronchoalveolar lavage were reduced 80% or more after 3 months of anti–IL-5 therapy numbers in bone marrow and bronchial biopsies were reduced 50% to 60%. This suggests that IL-5 antibody therapy effectively reduces circulating and airway luminal eosinophils, but less so bone marrow and lung parenchymal eosinophils.8 Cys-leukotrienes (LTs) may also play a role in eosinophil regulation.Circulating eosinophil counts in humans decrease by almost 20% after treatment with cyst-LT-receptor antagonists. These effects may reflect actions at the level of the bone marrow. Eosinophil–basophil colony-forming units increase in number, adhesion, and function, including production of IL-4 when exposed in vitro in the presence of LTD4. These responses are blocked by the presence of cysLT1 antagonists.
Activation of the humoral system occurs when naïve B lymphocytes with appropriate receptors (Igs) recognize an epitope in the intact foreign antigen. The antigen binds to the Ig, is ingested, and is processed such that it is expressed on the surface of the B cell in association with the same MHC (class I or II) that presented the antigen initially.4 Binding of the Ig receptor with the peptide and subsequent interaction with a CD4 cell that recognizes the antigen in its MHC complex stimulates proliferation of B cells, their differentiation into plasma cells, and the secretion of antibodies able to bind the epitope. Ig production is stimulated by IL-1, IL-4, IL-5, and IL-6 in B lymphocytes. The complete sequence occurs over 8 to 14 days and results in an anamnestic, or secondary, response. Generation of “memory” B and T cells provides a long-term mechanism for a rapid immune response on reexposure to the epitope (antigen).4
Soluble Components
Cytokines
More than 13 chemokine receptors are associated with rheumatoid arthritis, and at least 16 chemokines interact with these receptors.9 Both constitutive (responsible for physiologic trafficking and homing of the adaptive immune response) and inducible (responsible for effector white blood cell recruitment, including lymphocytes) responses are involved. A number of the chemicals also are responsible for angiogenesis. The inappropriate production of chemokines is associated with formation of an ectopic germinal center, which contributes to an uncontrolled immune response. Consequently, drugs directed toward chemokine receptors offer a therapeutic approach to control.
TNFα induces a broad spectrum of activity. Cytotoxicity of multiple cell types, including tumors, reflects both apoptosis and necrosis. A large number of proteins are targeted, along with other central mediators of the inflammatory process and immune activation. Examples include nuclear factors (e.g., NFκB), nitric oxide synthetase (NOS), cell-surface molecules, MHC classes I and II, and secreted proteins such as IL (e.g., -l, -6, -8), IFN, granulocyte-macrophage colony-stimulating factor, platelet-derived growth factor, urokinase plasminogen activator, and TNF-α itself. When administered exogenously, high concentrations of TNFα causes a toxic syndrome similar to septic shock. Bacterial lipopolysaccharide is the most potent stimulator of macrophage TNFα production. TNFα appears to mediate ischemia–reperfusion injury after transplantation of the liver, kidney, intestine, heart, lung, and pancreas and is a marker cytokine during organ rejection. Inhibitors have proved useful in human medicine for treatment of a number of autoimmune diseases, including Crohn’s disease, rheumatoid arthritis, psoriasis, and ankylosing spondylitis. Indications included corticosteroid-resistant graft-versus-host disease after bone marrow transplantation.10
Not surprisingly, the pervasive role of TNFα has led to the development of drugs that inhibit its actions. Included are glucocorticoids, pentoxifylline, and monoclonal immunoglobulin G (IgG) antibodies (e.g, infliximab, etanercept and the “humanized” adalimumab) etanercept, a humanized soluble TNFα receptor (TNFR) construct, and onercept, a TNFα–binding protein.10 A variety of other drugs targeting TNFα or its receptors are currently under investigation, as well as metalloproteinase inhibitors such as TNFα-converting enzyme (TACE). The use of these drugs, not surprisingly, is associated with a number of side effects. These include immunosuppresion, acute infusion reactions, delayed-type hypersensitivity reactions, autoimmune diseases, cardiovascular and neurologic adverse events, malignancies and lymphomas, and infectious complications.
Complement
The complement cascade is a major effector mechanism of the immune response. The cascade results in highly amplified events that interact with other physiologic cascades, including the coagulation pathway, kinin formation, and fibrinolysis.11 Consequences of complement activation include opsonization, the release of biologically vasoactive peptides, and cellular lysis. Cell-mediated cytotoxicity is implemented by cytotoxic T lymphocytes, NK cells and antibody-dependent cell-mediated cytotoxicity (ADCC), mediated by a variety of cells that express surface Fc receptors. Effector cells of ADCC include monocytes, neutrophils, eosinophils, selected cytotoxic T cells, and NK cells.
Immunoglobulins
Five classes of Igs are recognized in animals. IgM is the largest, forming up to 15% of the total Ig present. IgM exists as a monomer or a large polymeric form. IgM is responsible for the primary Ig response; in animals, for some infections, it is the only defense.11 The availability of five binding sites renders IgM efficient at antigen binding and agglutination, virus neutralization, and opsonization. IgM also is a potent activator of complement. Because it is such a large molecule, unless vascular permeability is altered, most IgM stays in circulation.
IgA is produced in submucosal lymphoid tissues and regional lymph nodes. After secretion outside of the cell, it travels to epithelial cells, where the secretory component of the IgA acts as a receptor, binding to IgA and stimulating its endocytosis. The two components are eventually exocytosed and attach to the mucosal surface, where they provide a protective component, neutralize toxins, adhere to bacteria and viruses, and interact with parasites.11
Each Ig molecule monomer comprises two heavy chains and two light chains attached by covalent bonds. The number of bonds varies with the Ig; the number of dimers varies with the class. A number of fragments (generated by enzymatic cleavage) can be described, including Fab, the antigen-combining fragment, and Fc, the crystallizable fragment, which binds to Fc receptors found on cells of the innatue immune system (e.g., NK cells, macrophages, neutrophils and mast cells).11
Mucosal Immunity
Induction of nasal mucosal tolerance can be manipulated, leading to mucosally induced immune therapy against selected infectious diseases (e.g, in Escherichia coli in human medicine). Mucosal tolerance has the advantage of inducing local and thus targeted Th1- or Th2- type immune responses, thus avoiding the negative sequelae of systemic cytokine injection. Mucosal adjuvants co-administered with antigens include cytokines (e.g., IL-1 and IL-12), or chemokines which promote specific CD4+ T helper cell cytokine responses (e.g., RANTES, lymphotactin, macrophage inhibitory protein–1 [MIP-1]). Therapeutic modulation of mucosal immunity generally targets unique Th cells and cytokine responses. For example, inflammatory bowel diseases (IBDs) appear to reflect failure of oral tolerance to luminal antigens, resulting in an imbalance of regulatory cytokines involving both Th1 and Th2 cell–mediated inflammation.12 Differences exist between orally and nasally induced immune responses. For example, aging more negatively influences GALT compared to NALT immunity. Nasal vaccines are more effective than oral vaccines in the promotion of protective immunity in the genitourinary tract. That these differences exist suggests that pharmacologic therapies might also be manipulated to locally target systemic effects or to limit therapy to local effects only, thus increasing benefits versus risks of treatment and that these therapies are likely to differentials, affect the different CIMS.
Antimicrobial and Host Defense Peptides
Antimicrobial peptides (AMPs, also called host defense peptides;) are evolutionarily ancient yet essential small cationic molecules found in animals, plants, and bacteria. Because they exhibit antimicrobial activity against a wide range of bacteria, fungi, and viruses, they are considered part of mucosal immunity. AMPs primarily act as cations, interacting with the anionic structure of and thus disrupting microbial membranes.13 Phosphatidylcholine in eukaryotic cell membranes is thought to be more positive compared to that in prokaryotic membranes, thus decreasing attraction to (or repelling) positively charged AMPs. However, in addition to antimicrobial actions, AMPs also serve as multifunctional mediators of immunity, inflammation, and wound repair. The importance of AMP or HDP in veterinary medicine has been reviewed by Linde and coworkers.14
Hypersensitivity Reactions and Cytokine Storm
An imbalance in the activities of CD4 Th1 and Th2 subsets may be responsible for the onset or exacerbation of immune-mediated diseases. Increased concentrations of Th1 (and decreased Th2) are associated with response to viral and fungal infections, whereas increased concentrations of Th2 (and decreased Th1) are associated with increased production of IgE and IgA antibodies. Autoimmune diseases are associated with a predominance (reflecting an imbalance) of Th1. Imbalances also have been associated with resistance to infectious disease and malignancy.15 In contrast, allergic inflammatory diseases (eg., atopy, inflammatory bowel disease, asthma) may reflect an imbalance that favors Th2.
Multiple novel mechanisms have been identified for their underlying role in the pathogenesis of immune-mediated diseases. Most, if not all, involve transcription factors, each providing potential current of future opportunities for manipulation. Among the prominent immune-mediated diseases being intensely studied in an attempt to understand the molecular mechanisms of immune mediated diseases are diabetes type 1, rheumatoid arthritis, multiple sclerosis, IBDs, psoriasis, and systemic lupus erythematosus (SLE).16
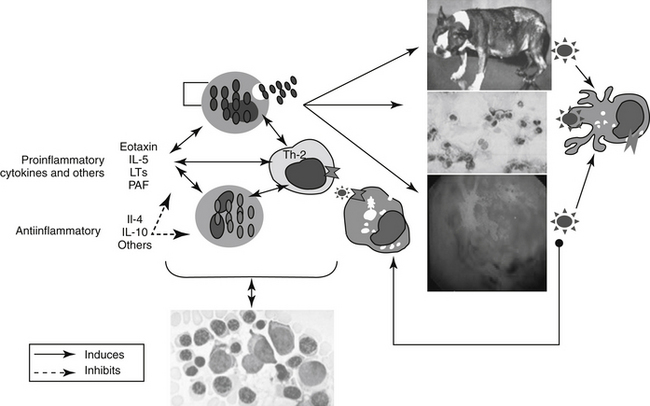
Four types of reactions result from activation of immunologic pathways. Type I hypersensitivity results from antigen–IgE interaction (Figure 31-2). IgE that has previously interacted with the antigen binds to the surface of a basophil or mast cell. Subsequent interaction with the same antigen causes mast cell degranulation and the release of a number of mediators associated with immediate hypersensitivity. Mediator release can be instantaneous (e.g., anaphylaxis); delayed for 2 to 4 hours; or biphasic, with both an immediate and a delayed reaction. Systemic release of mediator results in systemic anaphylaxis; localized mediator release limits reaction to the site of release (eg, swelling, redness, pain). Atopy is an inherited predisposition to develop IgE antibodies to environmental antigens and is characterized by constant high levels of IgE. An anaphylactoid reaction may be similar to anaphylaxis in presentation but reflects nonimmune-mediated mast cell degranulation (e.g., cationic drug-induced) (see Chapter 4).
Type II hypersensitivity occurs when ADCC occurs after antibody binds to a cell or to an exogenous antigen associated with a cell surface or a basement membrane. Complement may be activated and contribute to the damage. Examples of type II hypersensitivity include drug hypersensitivities, autoimmune hemolytic anemia, immune-mediated thrombocytopenia, immune-mediated endocrinopathies, and immune-mediated dermatologic disorders such as bullous pemphigus.11
Type III hypersensitivity results from the formation of antigen–antibody or immune complexes that either circulate or are deposited as microprecipitates in vascular beds or basement membranes. The Arthus reaction occurs 2 to 4 hours after IgG interacts with an antigen in the vessel wall. Serum sickness occurs when circulating immune complexes develop as a result of intravascular injection of the antigen. Microprecipitates in circulation deposit in basement membranes and the vascular endothelium, resulting in immune complex diseases. The risk of serum sickness increases with the persistence of antigen. The size of the immune complex also determines the degree of damage because larger complexes are more likely to deposit and initiate inflammation. Complement both contributes to and protects against damage caused by immune complex disease.11
Type IV hypersensitivity involves sensitized T cells that initiate a cell-mediated reaction on interaction with the appropriate class II MHC antigen. Lymphocyte and macrophage influx to the site occurs over a 24- to 72-hour period. Allergic contact dermatitis is an example of a type IV hypersensitivity.11
The term cytokine storm, or hypercytokinemia, has been used to describe the inappropriate systemic reaction resulting from the release of cytokines (more than 150) from a healthy and reactive immune system. Its importance emerged in response to the impact that swine flu to have in some, but not all afflicted human patients.17 The immune system appears to lose the control normally provided through a system of checks and balances involving both inflammatory signals (e.g., TNFα, IL-1, and IL-6) and antiinflammatory signals (e.g., IL-10), as well as coagulation factors, and oxygen free radicals. The impact of these signals on body tissues and organs can be profound and is exemplified by acute respiratory distress syndrome (particularly that associated with influenza or flu), sepsis, and systemic inflammatory response syndrome. Inhibiting T cell response has been proposed as a potential mechanism of treatment or prevention. Drugs that alter production or the impact of TNFα are among those studied for possible efficacy.
Regulation of the Immune Response Transcription Factors
Regulation of the immune response reflects a balance of the integrated actions of proinflammatory and antiinflammatory cytokines that act to trigger signaling pathways. The pathways, in turn, modulate gene expression program in the cells. Transcription factors targeted by cytokines include NFκB (proinflammatory),18 activator protein 1 (AP-1), SMAD proteins (responsible for transfer of extracellular signals from transforming growth factor to intracelluar nuclear TGF-ß gene transcription), glucocorticoid receptors (antiinflammatory)19,20 (see Chapter 30), and members of the STAT protein family (proinflammatory and antiinflammatory).5
NFκB is a transcription factor that, upon induction by a number of inflammatory agents, participates in the expression of a large number of target genes, many of which regulate both innate and adaptive immunity. Because a number of the target genes activate NFκB, signal amplification may occur at very low concentrations of the inciting antigen, causing profound effects. Regulatory failure has been associated with a number of autoimmune diseases, including IBDs such as Crohn’s disease and ulcerative colitis.18 NFκB appears to play a key role in rheumatoid arthritis in humans. Metalloproteinase-1(MMP-1) and TNF-α are among the molecules it regulates.16 The NFκB system is vital to survival, but regulatory activity is very complex, and undirected inhibition may result in undesirable immune suppression. For example, pharmacologic interference with a potentially therapeutic aspect (e.g., inactivation in enterocytes such that a systemic inflammatory response is avoided) often is accompanied by potentially lethal parallel effects (e.g., severe apoptotic damage to the reperfused intestinal mucosa). Pharmacologic manipulation is most likely to succeed only when tissue- or organ-targeted inhibition is possible. Another major role player is activator protein 1 (AP-1), a transcription factor with major proinflammatory effects. It regulates gene expression stimulated by cytokines, growth factors, stress, and bacterial and viral infections, thus controls diverse cellular processes such as differentiation, proliferation, and apoptosis.
Patients with glucocorticoid-sensitive versus glucocorticoid-resistant chronic inflammatory disease exhibit different cellular activation of NFκB, and AP-1 and upstream kinases; activation occurs in macrophages of the lamina propria in steroid-responsive patients but predominantly in epithelial cells of steroid resistant patients. Thus directed therapy for steroid-resistant patients might focus on inhibition of NFκB activation in epithelial cells.18
The Janus kinase (JAK)–STAT pathway is a major signaling pathway by which cytokine signals lead to the expression of genes that regulate immune cell proliferation and differentiation. The JAK–STAT signaling pathway is activated in response to cytokines (e.g., ILs and IFNs) as well as certain peptide hormones. The binding of the signaling molecules to the receptors causes activation (phosphorylation) of JAKs (e.g., JAK-1 through -3), which provide docking sites on the JAK protein for STAT proteins. The STAT proteins are then activated (phosphorylation) and translocated to the nucleus, where they bind to their response elements in the promoter of target genes. Transactivation cannot occur without co-activation by proteins such as acetyl transferase binding protein (e.g., CREB, a cyclic adenosine monophosphate [cAMP]–responsive element binding protein that increases MMP production in the synoviocytes of patients with rheumatoid arthritis).16
Cytokines that promote immune and inflammatory responses (e.g., IL-6, IFN-γ, IL-12, and IL-18) as well as those that suppress the immune response (e.g., IL-4, IL-10, IL-13) mediate cellular responses through the JAK–STAT signaling pathway. Up to seven STAT (including STAT-1 through -4, 5a and 5b, and STAT-6) proteins have been identified in mammals, each with a specific function in the immune response that modulates either proinflammatory or antiinflammatory responses. For example, STAT-3, originally discovered as an acute-phase response factor activated by IL-6, is also activated by many other cytokines and appears to be a constitutive protein that influences chronic inflammation. However, its embryonic deletion is lethal.5
Immunomodulatory Effects of Opioids
The potential of opioids to influence the immune system was first recognized close to 100 years ago when the effects of opium on phagocytic function were observed.21 Heroin addicts are more susceptible to infection. Rodent studies indicate increased mortality and morbidity rates when opioid-treated animals are exposed to infectious agents. The presence of opioid receptors on cells of the immune system has been recognized since the late 1970s.21,22 Although further studies are needed to fully elucidate the effect of opioids on immune function, thus far both the cellular and humoral immune reactions are believed to be affected.
Effects of opioids on the immune system are variable but are directed centrally, rather than peripherally, and involve primarily, but not exclusively, supraspinal mu receptors. Effects (inhibitory versus stimulatory) are dose dependent but occur at clinically relevant doses in animal models. Multiple central opioid receptors and endogenous opioids appear to be involved in complex immune responses. NK cytolytic activity and mitogen-stimulated T-cell proliferation are reduced by centrally administered morphine by way of mu receptors, whereas antibody production is both increased and decreased by met-enkephalin, which probably reflects interaction with multiple receptors. Effects can occur with single or multiple administration of exogenous opioids. Both the neuroendocrine system and the autonomic nervous system may serve as the efferent mechanisms mediating central opioid modulation of the immune system.21 Tolerance appears to develop to some of the immunomodulatory effects, but this remains controversial. The clinical implications regarding the role of opioids in modulating the immune response are not clear.
Immunomodulatory Effects of Vitamin D
Increasingly, vitamin D (1,25, dihydroxyvitamin D3) is recognized to have a noncalcemic role. Among the proposed activities is support of the immune system, particularly T cell–mediated immunity.23 Both T lymphocytes and macrophages are characterized by a high density of vitamin D receptors, particularly in immature immune cells and mature CD8 cells. Vitamin D compounds have been demonstrated in animal models to selectively immunosuppress, effectively preventing or modulating autoimmune diseases, including systemic lupus erythematosus, encephalomyelitis, rheumatoid arthritis, and IBD. Suppression or prevention of transplant rejection has been demonstrated in animal models. Mechanisms may include stimulation of the antiinflammatory mediators transforming growth factor beta 1 (TGF-β 1) and IL-4. These effects appear to occur without negatively affecting normal immune defense mechanisms. Therapeutic use may be limited by the advent of hypercalcemia, leading to investigations of noncalcemic analogs; indeed, some suggestion exists that hypercalcemia may be required for immunosuppression.
Immunomodulation in Viral and Neoplastic Diseases
Biologica l response modifiers are discussed in Chapter 32. Their primary indications are for treatment of viral or neoplastic disease or (generally their antagonism) for treatment of immune-mediated disease.
Viral Diseases
Biological response modifiers potentially offer a logical and unique approach to the treatment of viral diseases because (1) viruses are capable of immunosuppression and (2) the immune response is an important determinant in the host’s ability to overcome viral infection.24,25 Resistance against and recovery from viral infections in mammals depend on three components of the immune system. The mononuclear phagocytic (reticuloendothelial) system represents the first barrier to viral infections, but it is nonspecific and not always efficient. Sensitized T lymphocytes provide specific cell-mediated immunity, which is followed later by humoral events, either narrowly specific (antibodies) or nonspecific (IFN). Multiple interactions occur among these systems. Unfortunately, several systems also contribute to the pathogenesis of viral disease. Viruses may cause immunosuppression by several mechanisms: They may directly injure or impair all classes of lymphocytes; cause the production of soluble immunosuppressing chemicals (e.g., IFN, p15[E] of feline leukemia virus [FeLV]) damage all three cell systems by infection, or cause an imbalance in immunoregulation, thus leading to overactivity of suppressor T cells.
Neoplastic Diseases
Many tumors have surface antigens toward which specific antibodies can be directed. Biological response modifiers can alter response to tumor antigen at one or several of these points, depending on the drug. Some biological response modifiers augment or restore normal host effector mechanisms by acting as either a mediator or an effector of the antitumor response. Transformation of tumor cells may be decreased or maturation of tumor cells may be increased by biological response modifiers. Host tolerance of damage caused by cytotoxic chemotherapeutic drugs also may be increased by biological response modifiers. Finally, biological response modifiers have been used to alter patient response to FeLV viremia and thus the subsequent associated neoplasms.26,27
Immunomodifying Drugs
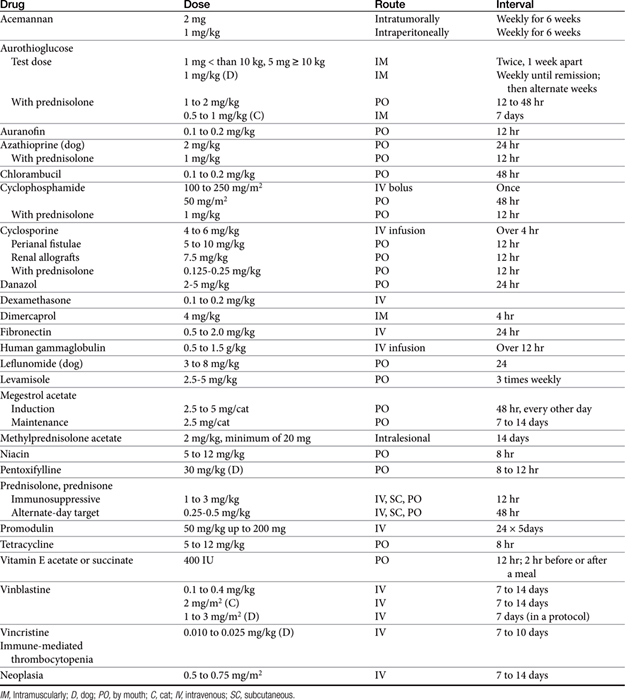
The sequelae of immunomodulation are not always beneficial; alteration of the immune response can impair many aspects of the host’s normal defense system. The use of immunomodulators remains in its early stages despite advances; this remains particularly true for veterinary medical applications. Because of the many facets of the immune system susceptible to regulation, the dose, timing, and route of administration of these drugs are important if undesired effects are to be avoided (Table 31-2). In addition, host effects such as age and nutritional status and the nature of the disease may be important determinants in the patient’s response to these drugs.
Marked advances have been made in the use of drugs that target the adaptive immune response. For the purposes of this text, drugs used to modify the immune response will include drugs whose actions are relatively nonspecific in their immunomoldulatory effect and those products of biological (animal) origin that target specific mediators of the immune response or their receptors, the latter is the focus of Chapter 32.
The role of P-glycoprotein in drug-induced immunomodulation is an emerging area of interest.28 The protein is expressed on peripheral blood mononuclear cells. It influences the secretion of cytokines secreted from antigen-presenting cells and selected T cells, and it has been shown to influence lymphocyte survival and antigen-presenting cell differentiation. These actions appear to contribute to the immunomodulatory actions of selected drugs capable of inhibiting P-glycoprotein (e.g., verapamil, progesterone, tamoxifen) and may account for the therapeutic immunomodulatory functions of these drugs. Eventually, these effects might be redirected for therapeutic benefit in allograft rejection and cell-mediated autoimmune disorders.
Immunosuppressant Drugs
Four major classes of immunosuppressive drugs are described in human medicine: glucocorticoids (discussed in Chapter 30), calcineurin inhibitors, antiproliferative–antimetabolic drugs, and biological agents.4 The latter includes biological response modifiers (Chapter 32).
Several principles should guide use of immunosuppressant drugs.4 First, suppression of the primary immune response is more easily accomplished than is suppression of the secondary (amnestic) response. Second, successful inhibition or suppression is easier if therapy begins before exposure to the inciting immunogen (antigen). Third, immunosuppressive drugs do not cause the same effect on all aspects of the immune system. Often, opposite effects are concentration dependent. Thus failure to achieve the desired response should not necessarily lead to an increase in dose. Finally, patients often require lifelong therapy, which increases the risk of adverse effects.
Two major limitations characterize immunosuppressive therapy. Patients receiving immunosuppressive therapy are predisposed to infections of any type. In addition, the risk of lymphomas and related malignancies is increased. This latter risk is more problematic in human patients because, in part, of their longer life span, but has proven a risk in dogs or cats receiving cyclosporine. Events that immunosuppressive drugs tend to target include antigen recognition, stimulation of IL-1, synthesis and release of IL-2 or other cytokines, and lymphocyte proliferation and differentiation.4 Secondary signal molecules are also becoming increasingly important as targets of immunosuppressive therapy.
Immunosuppressive drugs are used to treat immune-mediated disease, which in this chapter include autoimmune diseases (e.g., immune-mediated anemias or thrombocytopenias) as well as chronic allergic inflammatory diseases (e.g., atopic dermatitis and asthma, IBD). A third, less common indication in veterinary medicine is prevention of organ rejection in renal transplant patients. Autoimmune diseases are characterized by sensitization to endogenous proteins that are perceived to be foreign. Both cell-mediated and humoral responses can be directed toward the protein. Many immune-mediated disorders afflicting dogs and cats respond sufficiently well to glucocorticoids (discussed in Chapter 30) and, when necessary (in severe cases), cytotoxic drugs. These include immune-mediated autoimmune hemolytic anemia or thrombocytopenia, acute glomerulopathies, and many dermatologic disorders with an immune-mediated basis. However, the approval of cyclosporine (CsA) for chronic allergic dermatitis has increased its use for autoimmune diseases, providing an alternative to cytotoxic drugs as a second tier.
Cyclosporine A
Chemistry–Structure Relationship
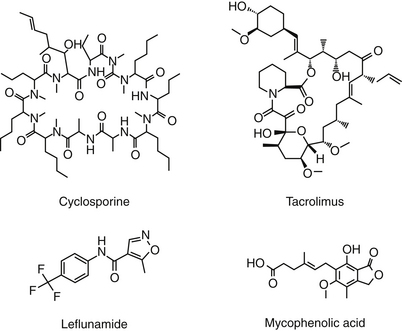
Cyclosporin A (CsA), now known as cyclosporine, is the most important immunosuppressive drug for human transplantation and the treatment of selective autoimmune disorders.4,30 CsA is one of nine cyclosporins (A through I), each a cyclic peptide drug (Figure 31-3) isolated from the fungi Cylindrocarpon lucidum and Trichoderma polysporum. An M designation, when present, indicates a metabolite. The drug is both very lipophilic (hydrophobic) and must be solubilized before administration.4 The oral preparation is a soft gelatin capsule (Sandimmune) or a (newer) microemulsion formulation (Neoral). The intravenous preparation is an ethanol–polyoxyethylated castor oil mixture. Historically, CsA has been formulated in peanut oil (for treatment of ophthalmic ocular disorders in dogs); however, an approved product formulated for topical use is now available.
Mechanism of Action
CsA is a unique immunosuppressant in that it specifically inhibits Th cells (both Th1 and Th2) early in their immune response to antigenic and regulatory stimuli4 without affecting suppressor cells. Suppression occurs as CsA binds to cyclophilin, a cytoplasmic receptor protein, forming a heterodimeric complex. This complex then binds to calcineurin. Binding of calcineurin inhibits calcium-stimulated phosphatase that dephosphorylates the regulatory protein. As such, CsA prevents transcription of T cell genes enhanced in response to T cell activation. Transcription mediated by IL-2, certain proto-oncogenes, and selected cytokine receptors are particularly affected.4 IL-2 production (and thus T cell proliferation and antigen-specific cytotoxic T lymphocyte generation) also is attenuated because the expression of TGF-β, a potent inhibitor of IL-2, is increased.4 B cells are not affected. The drug is most effective when administered before T cell proliferation has occurred. In addition to these immunomodulatory effects, CsA also affects other inflammatory cells. CsA inhibits skin mast cell numbers, survival, and response (secretory and histamine release) as well as secretion of IL-3, IL-4, IL-5, IL-8, and TNFα. Similarly, eosinophile response (release of granules and cytokines) and recruitment to allergic sites is impaired. CsA prevents TNFα-mediated late phase reactions, thus inhibiting IgE and mast cell–dependent cellular infiltration in the skin and bronchial mucosa. However, CsA does not inhibit IgA secretion or IgG and IgM formation in dogs and has no apparent effect on serum allergen-specific IgE levels, intradermal tests, or vaccination.
Surprisingly little information is available regarding immunomodulatory effects of CsA in dogs or cats. Using dermal microdialysis, Brazis and coworkers31 demonstrated that CsA at 5 mg/kg per day for 15 and 30 days decreased histamine, but not prostaglandin D2, release in sensitized Beagles challenged with Ascaris suum.
Preparations
CsA is available in a variety of preparations, two of which are approved in the United States for use in animals. CsA was first formulated for oral use in humans as vegetable oil. Oral absorption in this preparation depends on emulsification by bile acids. Poor oral bioavailability led to the formulation of a microemulsion preparation (modified CsA, ME), which, on contact with gastrointestinal fluids, disperses into a homogeneous monophasic microemulsion that mimics the mixed micellar phase of the standard formulation. In dogs the ME formulation offers a 35% bioavailability (Novartis Animal Health data on file).
Clinical Pharmacology
The pharmacokinetic behavior of CsA is complex, resulting in marked variability in the relationship between dose and blood concentrations.32 Variability reflects not only differences in both hepatic and intestinal metabolism (by CYP3A4) but also differences in P-glycoprotein among various tissues.32 The disposition of CsA has been well reviewed in the dog.33 Absorption after oral administration is slow and incomplete. The complex lipid nature of CsA complicates absorption by being dependent on bile acids, which generate a microemulsion. In humans oral bioavailability of the capsule ranges from 20% to 50% but is improved by 10% to 20% in the microemulsion form which is not dependent on bile acids.4,34 Peak concentrations occur 1 to 4 hours after oral administration in human beings. When the capsule—but not the microemulsion—is administered with a fatty meal, absorption is slowed. Studies suggest that decreased bioavailability of CsA after oral administration may reflect activity of drug-metabolizing enzymes of the intestinal epithelium35 or P-glycoprotein–mediated drug efflux.
KEY POINT 31-10
The kinetics of cyclosporine A are complicated by its large, liphophilic molecular structure.
The oral bioavailability of microemulsion in dogs is 35% compared with the 20% of the oral preparation.33 Bioavailability of orally administered CsA is approximately 25% to 30% after multiple oral administration in cats (n = 6). Food may impair the absorption of CsA; peak concentrations in fasted dogs were 20% higher than in nonfasted dogs in one study, although time to peak (approximately 1.5 hours) was the same.33 The drug is not predictably absorbed topically after transdermal administration (unpublished data, Boothe 2007). CsA is widely distributed, characterized by a large volume of distribution (13 L/kg in humans). The drug accumulates in erythrocytes (accounting for 50% or more of the drug in humans), and leukocytes (accounting for 10% to 20% of circulating drug in humans).4 Consequently, monitoring of whole blood rather than plasma or serum is recommended. Remaining circulating drug is bound to plasma lipoproteins. Further, concentrations in skin are up to tenfold higher than in blood, although this is based on homogenate data (as reviewed by Guaguère and coworkers33). In contrast, largely because of the influence of adenosine triphosphate–binding transporters P-glycoproteins, especially the ABCB1 cassette (P-glycoprotein, MDR1 gene product), little CsA crosses the blood–brain barrier.
CsA is metabolized to a large number of metabolites predominantly by the liver, although sufficient metabolism occurs by intestinal enterocytes that oral bioavailability is affected. Microflorae also may contribute to metabolism.37 CYP3A4 (commonly associated with P-glycoprotein or other efflux pumps) plays a major role in metabolism; many of the drug interactions involving CsA also involve CYP3A4 or associated glycoprotein. More than 25 to 30 metabolites have been documented in humans and dogs.32,38,39 Metabolism is targeted primarily toward the side chains rather than the ring structure and reflects hydroxylation, demethylation, sulfation, and cyclization. The major hydroxylated metabolites (AM1 and AM9) are further metabolized, contributing to the complex metabolic profile. Both AM1 and AM9 may contribute 10 to 20% of CsA activity, with AM1 contributing 27% of trough activity.40 In humans AM1 accumulates with chronic dosing and ultimately may surpass CsA, and monitoring assays might ideally detect the active compounds.39 They are detected to variable degrees by selected immunoassays based on a monoclonal antibody (e.g., Abbott TdX, Architect systems, Seimens Dimension).41,42 Selected laboratories may also offer a high-performance liquid chromatography (HPLC)–based system that, may or may not (generally not) quantitate the metabolites.39 Ultimately metabolism continues until the drug is totally inactivate the drug, although this has not been proved conclusively.4 Metabolites are excreted in bile and feces, with less than 2% of unchanged drug eliminated in the kidneys of dogs. CYP3A4 is largely responsible for metabolism.
The half-life of CsA in dogs is quite variable, depending on the investigator and assay. In blood the half-life ranges from a low of 5 hours (HPLC) to a high of 18 hours (using fluorescent polarized immunoassay [FPIA]); 29 hours was reported for serum.43 That reported for Atopica, the product approved for use in dogs, is 4.5 hours, a length more consistent with that measured in the author’s therapeutic drug monitoring laboratory using FPIA. In a manufacturer-sponsored affinity colony-mediated immunoassay (ACMIA) study, Steffan and coworkers44 reported the disposition of CsA (5 mg/kg) after single-dose oral administration of either capsules or solution in fasting or fed conditions using a randomized crossover design with a 1-week washout between studies (Table 31-3); food decreased peak concentrations and area under the curve by 22%.
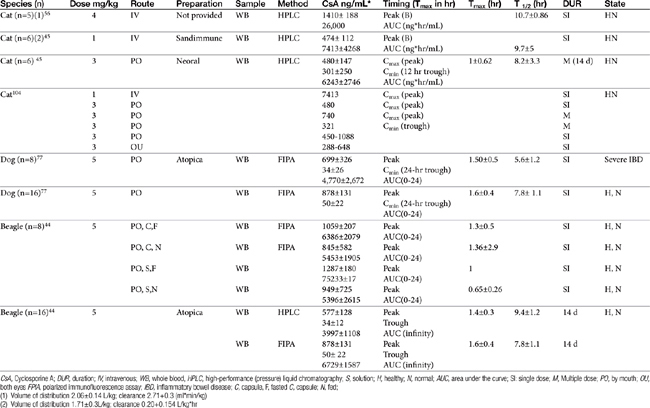
Mehl and coworkers45 reported the disposition of CsA in cats (n = 6 male) after single-dose intravenous (1 mg/kg) and multiple-dose (14 days; 3 mg/kg twice daily) as Neoral (see Table 31-3). Variability in plasma drug concentrations among cats and across time underlines the importance of monitoring if treating life-threatening conditions. After multiple-day (7 to 14 days) oral administration (3 mg/kg), mean (peak) plasma concentrations at 2 hours were 740 ± 326 (7 days) and 655 ± 285 ng/mL (14 days), whereas trough concentrations were 332 ± 237 and 301.5 ± 250 ng/mL, respectively. Oral bioavailability was only 25% to 29%.
Because cats may find the oral preparation unpalatable, the oral solution can be given diluted in olive oil. The intravenous preparation (4 to 6 mg/kg as a 4-hour intravenous infusion) can be given to animals for which oral administration is not possible.46 Ocular administration might serve as an alternative route for systemic delivery of CsA in cats.47 After ocular administration of either an oral preparation or an ocular preparation of CsA in olive oil, (5 mg/kg in each eye as a 10% solution) peak concentrations of 450 to 1033 ng/mL (oral preparation) and 288 to 648 ng/mL (ocular) were achieved, with an absorption lag time ranging from 0 to 1.34 hours for the oral solution and 0.27 to 1.2 hours for ocular preparation. The elimination half-life ranged from 2.41 to 10.04 hours (oral), and 3.09 to 15.75 hours (ocular), respectively. Blood concentrations were sufficient to inhibit lymphocyte activity as measured in vitro, leading the authors to conclude that ocular administration of CsA in an olive oil vehicle might be a reasonable alternative to oral, or even intravenous, administration in cats intolerant to the latter.
Drug Interactions
Novartis48 provides a transplant drug interactions monograph that cites more than 600 references and delineates cyclosporine interactions involving more than 300 drugs. The list of drugs that might interact with CsA is extensive. The manual is available on the Novartis website at no charge and should be consulted when treating patients receiving additional drugs. Selected interactions can be found in Table 31-4. This review will focus on some of the more common interactions, including those used intentionally.
Table 31-4 Cyclosporine Drug Interactions
| Drug | Impact |
|---|---|
| ACE inhibitors | Increased nephrotoxicity |
| Acyclovir | Falsely increased CsA |
| Allopurinol | Increased toxicity |
| Aluminum | Increased nephrotoxicity and CsA bioavailability |
| Aminoglycosides | Increased nephrotoxicity |
| Amlodipine | Increased gingival hyperplasia |
| Antacids | Increased aluminum toxicity |
| Azithromycin | Increased gingival hyperplasia |
| Increased CsA | |
| Bile acids | Increased bioavailability |
| Chloramphenicol | Decreaesed clearance, increased CsA |
| Cimetidine | Delayed absorption |
| No effect (?) on metabolism or absorption | |
| Ciprofloxacin | Antagonizes CsA immunosuppression |
| Cisapride | Increased absorption |
| Clarithromycin | Decreased clearance, increased CsA |
| Cyclophosphamide | Increased hepatotoxiciy |
| Danazol | Decreasd metabolism, increased absorption |
| Dexamethasole | Increased CsA metabolism; synergistic immunosuppression |
| Digoxin | Increased digoxin toxicity |
| Diltiazem | Increased gingival hyperplasia, increased CsA |
| Erythromycin | Increased hepatotoxicity (decreased metabolism?) |
| Famotidine | Decreased clearance, increased CsA |
| Fluconazole | Oral: increased bioavailability |
| Fluoxetine | Decreased clearance, increased CsA |
| Furosemide | Decreased nephrotoxicity |
| Glucocorticoids | Increased metabolism, decreased CsA |
| Itraconazole | Decreased clearance, increased CsA |
| Increased absorption(?) | |
| Ivermectin | Increased neurotoxicity |
| Ketamine | Combined proconvulsant effects |
| Ketaconazole | Decreased clearance, increased CsA |
| Increased absorption | |
| Increased toxicity | |
| Leflunomide | Synergistic immunosuppression |
| Loperamide | Altered absorption |
| Methotrexate | Decreased clearance, increased CsA |
| Increased methotrexate toxicity | |
| Metoclopramide | Increased absorption, increased CsA |
| Metoprolol | Beneficial hemodynamics |
| Mitoxantrone | Increased mitoxantrone concentrations |
| Mycophenolic acid | Synergistic immunosuppression |
| Norfloxacin | Decreased CsA clearance? |
| Omeprazole | Delayed CsA absorption, decreased CsA clearance? |
| Pancuronium | Prolonged duration of pancuronium effect |
| Pentoxifylline | Synergistic inhibition of TNF and other immunosuppression |
| Phenobarbital | Increased CsA clearance, decreased CsA |
| Phenytoin | Increased CsA clearance, decreased CsA |
| Prazosine | Increased afterload |
| Prednisone | Increased risk of hepatotoxicity, hyperlipidemia |
| Primidone | Increased CsA clearance, decreased CsA |
| Propranolol | Antagonism of CsA immunosuppression |
| Decreased propranolol clearance | |
| Ranitidine | Increased risk of hepatotoxicity, thrombocytopenia |
| Rifampin | Increased CsA clearance, decreased CsA |
| Sirolimus | Synergistic immunosuppression |
| Synergistic risk of toxicity | |
| Spironolactone | Hyperkalemia |
| Sucralfate | Increased aluminum |
| Sulfadiazines | Interference with HPLC assay results in increased CsA |
| Tacrolimus | Decreased CsA clearance, increased CsA |
| Terbinafine | Inceased CsA metabolism |
| Ticlopidine | Altered CsA metabolism |
| Trimethoprim | Increased CsA clearance, decreased CsA |
| Ursodeoxycholic acid | Decreased Tmax |
| Vasopressin | Enhanced vasopressin effects |
| Verapamil | Gingival hyperplasia, synergistic immunosuppression |
| Vitamin D3 | Additive suppressive effects |
| Voriconazole | Decreased metabolism? |
ACE, angiotensin-converting enzyme; CsA, cyclosporine A.
From Neoral and Sandimmune Drug Interactions, Novartis package insert, 2007
The enzyme that metabolizes CsA (CYP3A4) is responsible (in humans) for approximately 30% of all drug metabolism; many of CsA drug interactions occur with this enzyme. Further, CsA is a target of P-glycoprotein, another site characterized by a substantial number of CsA–drug interactions.4,49 The risk of drug interactions is another indication of the need for therapeutic drug monitoring as a guide to proper dosing regimens. CsA elimination is accelerated, probably in part because of the induction of drug-metabolizing enzymes, by phenytoin, phenobarbital, rifampin, and sulfamethoxazole–trimethoprim combinations, and others. Dexamethasone also is an inducer of CsA.32 Its elimination is decreased by amphotericin B, erythromycin, and ketoconazole.4 CsA also appears to alter the oral absorption of other drugs.35 CsA use with other immunosuppressive drugs may benefit from these interactions, including glucocorticoids.4
The inhibitory effect of ketoconazole on intestinal epithelial and hepatic drug metabolism and on P-glycoprotein efflux may be of therapeutic benefit in patients receiving CsA by decreasing hepatic clearance and increasing oral bioavailability.35,50–53 Ketoconazole may also decrease the lipoprotein that binds CsA, resulting in higher concentrations of free CsA.51 Studies with ketoconazole in humans found the dose of CsA to be reduced by approximately 70% to 85%.54 The use of ketoconazole in conjunction with CsA as a means of decreasing drug cost has been well accepted in human transplant patients.51 The two drugs have been used safely for up to 47 months in one study reducing the CsA dose as much as 88%.51 Although the effects emerge rapidly, with 62% of the effect is apparent by day 7, the maximum inhibitory effect may not be present for 12 months. This has implications regarding monitoring and supports the need for collection of peak and trough samples such that the impact of ketoconazole on drug elimination half-life might be determined.
The effect of combining ketoconazole with CsA has been studied in dogs.55 Ketoconazole was studied at doses ranging from 1.25 to 20 mg/kg per day, with the magnitude of inhibition increasing with the dose of ketoconazole above but not below 2.5 mg/kg.55 Clearance was reduced by 85% at 10 mg/kg per day. Differences in clearance did not result in significant differences in oral bioavailability. In their review, McNaulty and Lensmeyer56 indicated that in dogs, CsA half-life will increase over twofold and decrease the concentration of active metabolites in response to 2.5 to 10 mg/kg ketoconazole. Up to 75% of the CsA dose could be reduced experimentally in Beagles concurrently receiving a dose of ketoconazole of 13.6 mg/kg per day.57 In a study in dogs with perianal fistulae, Mouatt58 examined the impact of ketoconazole (10 mg/kg once daily) on CsA concentrations (1 mg/kg twice daily). Samples were collected 12 hours after the last dose (duration not clear). Data were not provided on all dogs, but the author noted that at 1.1 mg/kg twice daily of CsA, all dogs exceeded 200 ng/mL at through (12 hours); nine dogs exceeded 900 ng/mL, and two dogs exceeded 1500 ng/mL. Concentrations varied by 10% to 40% in the same dog despite no dose change. This may have reflected variability in time to steady state, which took 2 to 4 weeks. A trough concentration of 200 ng/mL was targeted. Although 8 dogs maintained concentrations of 220 to 520 ng/mL with CsA doses as little as 0.35 to 0.55 mg/kg twice daily, one dog required only 0.16 mg/kg twice daily to maintain 159 to 225 ng/mL, whereas three others required 0.95 to 1.1 mg/kg twice daily to maintain 120 to 235 ng/mL. This marked variability in response to ketaconozole appears to occur in clinical patients as is suggested by samples submitted to the author’s therapeutic drug monitoring laboratory. Marked variability occurs not only in concentrations, but in the time to steady-state, which in turn determines when monitoring should be implemented. For example, the author’s laboratory has demonstrated prolongation of the half-life of CsA in a patient from 4 hours (as reported in normal dogs) to over 150 hour. Steady state was not achieved in this patient until approximately 4 weeks, causing CsA concentrations to continue to increase despite a decrease in dose that was based on monitoring at 1 week. Yet, in other patients, half-life has been less than 12 hours, despite ketoconazole therapy. Detecting these diffrences requires collection of peak and trough samples such that half-life and time to steady-state can be determined in each patient.
The simultaneous administration of ketoconazole with CsA also has been studied in cats. McNaulty and Lensmeyer56 prospectively studied the impact of a two doses of ketoconazole (10 mg/kg at 24 and 0.5 hr) before a single dose of CsA (4 mg/kg intravenously) in cats (n = 5) using a randomized crossover design with a 14-day washout. Concentrations of CsA were detected using HPLC. Clearance was reduced from 2.73 ± 0.3 to 1.22 ± 0.18 mL∗kg/min, resulting in an elimination half-life prolongation from 10 ± 0.9 to 21 ± 3 hours.
Several studies have examined the impact of cimetidine, an inhibitor of CYP3A4, CYP2D6, and CYP1A2 on CsA concentrations. Cimetidine also is a substrate for P-glycoprotein. The results of the studies are contradictory, perhaps reflecting differences in duration of therapy and species. D’Souza and coworkers59 found that 5 days of cimetidine therapy significantly decreased CsA clearance, increased volume of distribution (perhaps by impacting P-glycoprotein in distribution tissues) and prolonged elimination half-life in rabbits, whereas Bar-Meir and coworkers60 found that short-term administration of cimetidine had no effect on CsA metabolism in rats. Shaefer and coworkers61 found that 7 days of dosing with cimetidine or famotidine did not affect CsA pharmacokinetics in healthy men. Daigle62 explored the impact of cimetidine (15 mg/kg orally every 8 hours for 8 days) on the disposition of CsA (5 mg/kg orally per day; the last 3 days of cimetidine administration) in dogs using a randomized crossover design with a 14-day washout. Significant differences could not be demonstrated for Cmax or area under the curve the power to detect a significant change was not addressed. The author’s therapeutic drug monitoring laboratory has identified some patients for which a cimetidine–CsA interaction may be present, but, other patients for which no effect could be measured.
Among the other drugs that impair CsA elimination or compete with efflux pumps include itraconazole, the calcium channel blockers such as diltiazem, and the macrolide antibacterial erythromycin.50 In humans, diltiazem decreases the CsA dose by approximately 30% to 50%.54 Other drugs that influence CYP activity should be expected to potentially affect CsA. For example, in the author’s laboratory, a cat receiving azithromycin (5 mg/kg once daily) along with CsA (5 mg/kg twice daily), exhibited peak concentrations exceeded 4500 ng/mL, presumably in part resulting from an elimination half-life that exceeded 150 hours. Azithromycin also competes for P-glycoprotein. Other drugs that induce P-glycoprotein might decrease CsA concentrations. For example, P-glycoprotein is upregulated in the duodenum of dogs receiving glucocorticoids.62a
Side Effects
CsA is characterized by a narrow therapeutic index in human patients, with renal toxicity the primary adverse effect. Renal tubular cells develop hyperuricemia (worsened by diuretics) and hyperkalemia (a renal tubular and erythrocyte ion channel effect).63 Hepatic injury also occurs. Although less common than renal dysfunction, the risk of severe hepatic damage is markedly increased when CsA is used in combination with cytotoxic drugs. CsA also increases the incidence of gallstones in human patients. However, in contrast to humans, renal and hepatic toxicities do not appear to be in dogs and cats. Risk factors, such as concurrent administration of nephroactive or nephrotoxic drugs, or renal transplantation have not been addressed in dogs or cats. Hyperlipidemia also has been a reported side effect in humans, particularly in patients receiving glucocorticoids. Other side effects reported in humans include neurotoxicity, gastrointestinal upset, and hypertension.4 Development of B cell lymphoma also has been reported in humans; an incidence 10% has been reported in cats undergoing renal transplantation, with mean time to onset of 9 months. 63a Side effects in dogs include gastrointestinal upset and dermatologic or mucosal abnormalities. Vomiting may occur in up to 40% of dogs, although it may be intermittent and short in duration. Diarrhea occurs less commonly (16% to 18%).Vomiting may be more likely when CsA is combined with ketoconazole, but this may reflect higher CsA concentrations. Dermatologic abnormalities reported in dogs include hair loss (possibly reflecting hair growth and pushing of hair from follicles), gingival hyperplasia, and gingivitis in up to 33% of dogs.58 In one study,58 hair loss resolved by 7 weeks, although hairy coats were thinner at study end (see Therapeutic Use). Hyperplasia may reflect an imbalance of fibroblast formation and collagen degradation by collagenase, although stimulation of TGF-β also has been suggested.33 Stimulation of TGF-β also stimulates extracellular matrix and decreases degrading proteases. Treatment with antimicrobials (metronidazole, spiramycin) may be useful for treating hyperplasia.58 Skin healing might be supported rather than inhibited, as occurs with glucocorticoids.67
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


