Chapter 3 HYPOTHYROIDISM
PHYSIOLOGY OF THE THYROID GLAND
THYROID HORMONE SYNTHESIS.
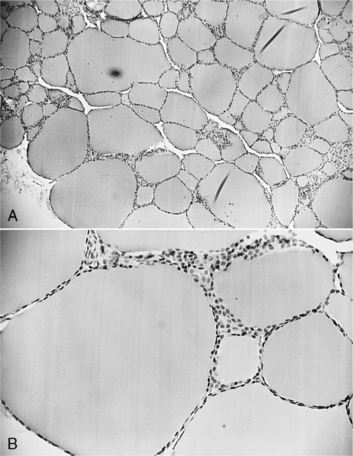
The basic functional unit of the thyroid gland is the follicle, a hollow sphere of cells surrounded by a basement membrane (Fig. 3-1). The wall of the follicle is a single layer of thyroid cells, which are cuboidal when quiescent and columnar when active. The follicular lumen contains colloid, a viscous gel that is primarily a store of thyroglobulin secreted by the thyroid cells. Thyroglobulin is a large glycoprotein dimer containing iodotyrosines that serve as precursors for thyroid hormone synthesis.
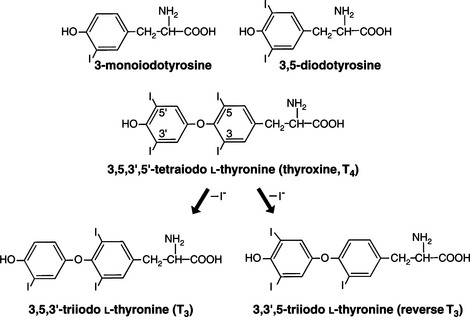
Adequate ingestion of iodide is a prerequisite for the normal synthesis of thyroid hormones by the thyroid. Iodide is actively transported from the extracellular fluid into the thyroid follicular cell, where it is rapidly oxidized by thyroid peroxidase into a reactive intermediate that is then incorporated into the tyrosine residues of acceptor proteins, primarily thyroglobulin (Greenspan, 2001). Iodinated tyrosine residues (monoiodotyrosine [MIT], diiodotyrosine [DIT]) in thyroglobulin combine to form iodothyronines—thyroxine and triiodothyronine (Fig. 3-2).
Thyroglobulin is stored extracellularly in the follicular lumen. As a prerequisite for thyroid hormone secretion into the blood, thyroglobulin must first reenter the thyroid cell and undergo proteolysis. Pseudopods from the apical cell surface extend into the colloid in the follicular lumen, and large colloid droplets enter the cytoplasm by endocytosis (Greenspan, 2001). Each colloid droplet is enclosed in a membrane derived from the apical cell border. Electron-dense lysosomes then fuse with the colloid droplets to produce phagolysosomes. These phagolysosomes migrate toward the basal aspect of the cell, during which time lysosomal proteases hydrolyze thyroglobulin. Thyroxine and, to a much lesser degree, triiodothyronine liberated from thyroglobulin by the proteolytic process pass from the phagolysosome into the blood, possibly by diffusion. Most of the liberated iodotyrosines (MIT, DIT) are deiodinated, thus releasing iodide, which can be reused for thyroglobulin iodination or can diffuse out into the circulation. A small quantity of intact thyroglobulin also enters the circulation. This leakage can be increased when the thyroid cells are damaged, such as occurs with lymphocytic thyroiditis.
REGULATION OF THYROID FUNCTION.
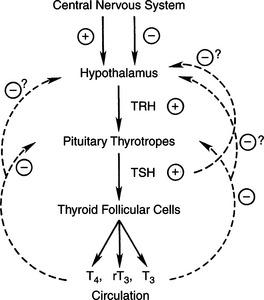
Thyroid hormone synthesis and secretion are regulated by extrathyroidal (thyrotropin) and intrathyroidal (autoregulatory) mechanisms. Thyrotropin (TSH) is the major modulator of thyroid activity, the net result of which is increased thyroid hormone secretion (Fig. 3-3). TSH secretion by the pituitary is modulated by thyroid hormone in a negative feedback regulatory mechanism. At the pituitary, it is primarily 3,5,3’-triiodothyronine (T3), produced locally by the monodeiodination of thyroxine (T4), that inhibits TSH secretion (Greenspan, 2001). The “thermostat” setting of the thyroid hormone–TSH feedback loop is modulated by thyrotropin-releasing hormone (TRH) from the hypothalamus. Hypothalamic production and release of TRH are controlled by poorly understood neural pathways from higher brain centers.
THYROID HORMONES IN PLASMA.
Thyroid hormones in plasma are largely bound to protein, most notably thyroid hormone–binding globulin (TBG), thyroxine-binding prealbumin (TBPA), albumin, and certain plasma lipoproteins. Less than 1% of T4 and T3 circulate “free” in the unbound state. Free or unbound thyroid hormones enter cells, produce their biologic effects, and are in turn metabolized. Only the free hormone regulates the pituitary feedback mechanism. Protein-bound thyroid hormones serve as a large reservoir that is slowly drawn upon as the free hormone dissociates from the binding proteins and enters the cells. Thyroid hormone secretion is precisely regulated to replenish the metabolized thyroid hormones.
THYROID HORMONE METABOLISM.
Thyroxine is the major secretory product of the normal thyroid. The major pathway of T4 metabolism is the progressive deiodination of the molecule. The initial deiodination of T4 may occur in the outer ring, producing T3, or in the inner ring, producing reverse T3 (rT3; Fig. 3-2). Because conversion of T4 to T3 represents a “step up” in biologic activity, whereas conversion of T4 to rT3 has the opposite effect, the conversion of T4 to T3 or rT3 by outer or inner ring iodothyronine deiodinase is a pivotal regulatory step in determining thyroid hormone biologic activity. Less than 20% of total T3 is produced in the thyroid, and the remaining 80% to 90% is derived from outer ring monodeiodination of T4 in peripheral tissues. The tissues that concentrate the most thyroid hormone are liver, kidney, and muscle (Greenspan, 2001). Conjugation of thyroid hormone to soluble glucuronides and sulfates with subsequent excretion in the bile and urine represents another major metabolic pathway for thyroid hormone.
THYROID HORMONE ACTIONS.
The thyroid hormones affect many metabolic processes, influencing the concentration and activity of numerous enzymes; the metabolism of substrates, vitamins, and minerals; the secretion and degradation rates of virtually all other hormones; and the response of their target tissues to them. Thyroid hormones are critically important in fetal development, particularly of the neural and skeletal systems. Thyroid hormones stimulate calorigenesis, protein and enzyme synthesis, and virtually all aspects of carbohydrate and lipid metabolism, including synthesis, mobilization, and degradation (Larsen et al, 1998). Furthermore, thyroid hormones have marked chronotropic and inotropic effects on the heart; are necessary for normal hypoxic and hypercapnic drive to the respiratory centers; stimulate erythropoiesis; and stimulate bone turnover, increasing both formation and resorption of bone (Greenspan, 2001). In essence, no tissue or organ system escapes the adverse effects of thyroid hormone excess or insufficiency.
CANINE HYPOTHYROIDISM
CLASSIFICATION
Structural or functional abnormalities of the thyroid gland can lead to deficient production of thyroid hormone. A convenient classification scheme is based on the location of the problem within the hypothalamic– pituitary–thyroid gland complex (Fig. 3-3). Primary hypothyroidism is the most common form of this disorder in the dog, resulting from problems with-in the thyroid gland. In the dog, destruction of the thyroid gland is the usual cause of primary hypothyroidism. Congenital defects in hormonogenesis have also been documented but are rare. Secondary hypothyroidism follows dysfunction within the pituitary thyrotropic cells, causing impaired secretion of TSH and a “secondary” deficiency in thyroid hormone synthesis. Secondary hypothyroidism, rare to uncommon in dogs, could follow destruction of pituitary thyrotrophs (e.g., pituitary neoplasia) or suppression of thyrotroph function by hormones or drugs (e.g., glucocorticoids). Tertiary hypothyroidism is defined as deficient TRH secretion by peptidergic neurons in the supraoptic and paraventricular nuclei of the hypothalamus. Tertiary hypothyroidism has not been reported in dogs and can be assumed to be extremely rare.
Disorders causing secondary or tertiary hypothyroidism would result in thyroid gland atrophy and no real “pathology.” The thyroid gland remains potentially responsive to TSH or TRH administration, although the response may be absent or suppressed with chronic lack of stimulation from the pituitary gland. In contrast, progressive pathologic lesions within the thyroid are usually identifiable in dogs with primary disease. Thyroid responsiveness to TSH and TRH becomes severely impaired after significant primary disease. This failure of responsiveness of the thyroid gland to TSH or TRH has historically been used to diagnose the disorder.
ETIOLOGY
Primary Hypothyroidism
Primary hypothyroidism is the most common cause of naturally occurring thyroid failure in the adult dog, accounting for more than 95% of our hypothyroid cases. Two histologic forms of primary hypothyroidism predominate in the dog: lymphocytic thyroiditis and idiopathic atrophy (Table 3-1). The end result of both these forms is the same—progressive destruction of the thyroid gland and resultant deficiency of circulating thyroid hormones.
TABLE 3-1 POTENTIAL CAUSES OF HYPOTHYROIDISM IN THE DOG
* Established etiology in the dog
LYMPHOCYTIC THYROIDITIS.
Lymphocytic thyroiditis is characterized histologically by a diffuse infiltration of lymphocytes, plasma cells, and macrophages within the thyroid gland, resulting in progressive destruction of follicles and secondary fibrosis (Gosselin et al, 1981a; Fig. 3-4). Neutrophils are usually not abundant and, when present, are associated with necrotic areas of the thyroid gland. Destruction of the thyroid gland is progressive, and clinical signs may not become evident until more than 75% of the gland is destroyed. Based on experiences following dogs with positive thyroglobulin autoantibody test results (see page 124), the onset of clinical signs and development of decreased serum thyroid hormone and increased serum TSH concentrations is usually a gradual process, often requiring 1 to 3 years to develop, suggesting that the destructive process is slow (Nachreiner et al, 2002). Graham et al (2001a) have proposed several stages in the development of lymphocytic thyroiditis in dogs, beginning with a subclinical stage characterized by positive thyroglobulin and thyroid hormone autoantibody tests and minimal inflammation in the thyroid gland through progressively worsening inflammation and destruction of the thyroid gland and the eventual development of increased serum TSH and decreased serum thyroid hormone concentrations (Table 3-2). The terminal stages of destruction are characterized by few follicles and extensive adipose connective tissue replacement of atrophied parenchymal structures, with residual infiltrates of lymphocytes and plasma cells interspersed with nests of parafollicular cells and a few small follicles containing degenerating follicular cells and poorly staining colloid (Conaway et al, 1985b). Analysis of age distributions of dogs with laboratory test results (i.e., thyroglobulin antibody, T4, and TSH) consistent with the different stages or classifications of lymphocytic thyroiditis suggest that the age of peak prevalence progresses by 1 to 2 years through each of the classifications (Graham et al, 2001a). Studies also suggest that differences in progression rate or likelihood of progression may exist for different breeds of dogs.
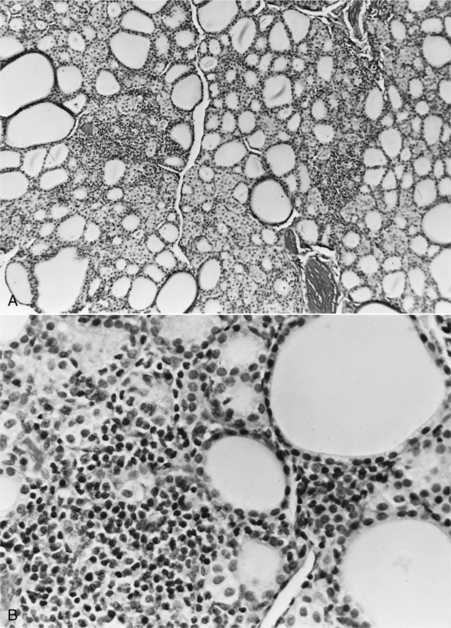

Lymphocytic thyroiditis is an immune-mediated disorder. Involvement of humoral immune mechanisms is supported by these findings: increased incidence of circulating autoantibodies to thyroid antigens, including thyroglobulin, T4, and T3; identification by electron microscopy of thickened basement membranes containing electron-dense deposits, which are believed to be antigen-antibody complexes, in thyroid follicles; and the induction of lesions similar to lymphocytic thyroiditis in dogs following the intrathyroidal injection of thyroglobulin antibodies (Gosselin et al, 1980; 1981a; 1981b; 1981c; Gaschen et al, 1993). Antibody binding to the follicular cell, colloid, or thyroglobulin antigens is believed to activate the complement cascade, antibody-dependent cell-mediated cytotoxicity, or both, causing follicular cell destruction. Identifiable thyroid antigens include the following: thyroglobulin, which is the main antigen in colloid and has the strongest correlation with the presence of lymphocytic thyroiditis; colloidal antigen (CA-2), a nonthyroglobulin, noniodine component of colloid; microsomal antigen and antinuclear substance antigen, located within the follicular cells; and cell-surface antigen (Gosselin et al, 1980; Vajner, 1997).
The cell-mediated immune system may also play an important, possibly primary, role in the development and perpetuation of lymphocytic thyroiditis. A defect in suppressor T cell function is suspected in this disorder. This would allow effector T lymphocytes to attack follicular cells and allow helper T cells to induce plasma cell differentiation, with subsequent production of thyroid antibodies (Strakosch et al, 1982).
The initiating factors involved in the development of lymphocytic thyroiditis are poorly understood. Genetics undoubtedly plays a major role, especially given the increased incidence of this disorder in certain breeds and in certain lines within a breed (see page 96). Lymphocytic thyroiditis is an inherited disorder in colony-raised Beagles, with a polygenic mode of inheritance, and was identified as an autosomal-recessive trait in a family of Borzoi dogs (Conaway et al, 1985a). An increased frequency of circulating thyroid hormone autoantibodies has also been found in certain breeds (Table 3-3; Nachreiner et al, 2002). Environmental risk factors have not been well defined in the dog. A link between infection and development of lymphocytic thyroiditis has been speculated but not proved (Penhale and Young, 1988). Infection-induced damage to the thyroid gland, causing release of antigens into the circulation and their subsequent exposure to the host’s immune system, or antigenic mimicry of thyroid antigens by viral or bacterial agents could initiate the immune-mediated inflammatory process. Vaccine administration has also been hypothesized to be a contributing factor for development of lymphocytic thyroiditis (Smith, 1995; Allbritton, 1996). A recent study documented a significant increase in antibovine and anticanine thyroglobulin antibodies in research Beagles after repeated vaccinations beginning at 8 weeks of age (Scott-Moncrieff et al, 2002). A significant increase in anticanine thyroglobulin antibodies was also documented in adult pet dogs 2 weeks after vaccination. The clinical importance of this finding as it relates to development of lymphocytic thyroiditis is unknown. The study did not determine how long the thyroglobulin antibodies persisted, although antibody levels had decreased to prevaccination values by the time of the next vaccination in most research dogs. None of the dogs in the study, with the exception of three dogs believed to have developed spontaneous thyroiditis, developed evidence of thyroid dysfunction by 4.5 years of age. The authors speculated that the antithyroglobulin antibodies detected in their study was the result of contamination of the vaccine by bovine thyroglobulin. There is considerable sequence homology for thyroglobulin between species, and antibodies to thyroglobulin have some species cross-reactivity (Tomer, 1997).
TABLE 3-3 DOG BREEDS REPORTED TO HAVE AN INCREASED PREVALENCE OF THYROID HORMONE AUTOANTIBODIES
| Breed | Odds Ratio* |
|---|---|
| Pointer | 3.61 |
| English Setter | 3.44 |
| English Pointer | 3.31 |
| Skye Terrier | 3.04 |
| German Wirehaired Pointer | 2.72 |
| Old English Sheepdog | 2.65 |
| Boxer | 2.37 |
| Maltese | 2.25 |
| Kuvasz | 2.18 |
| Petit Basset Friffon Vendeen | 2.16 |
| American Staffordshire Terrier | 1.84 |
| Beagle | 1.79 |
| American Pit Bull Terrier | 1.78 |
| Dalmatian | 1.74 |
| Giant Schnauzer | 1.72 |
| Rhodesian Ridgeback | 1.72 |
| Golden Retriever | 1.70 |
| Shetland Sheepdog | 1.69 |
| Chesapeake Bay Retriever | 1.56 |
| Siberian Husky | 1.45 |
| Brittany Spaniel | 1.42 |
| Borzoi | 1.39 |
| Australian Shepherd | 1.28 |
| Doberman Pinscher | 1.24 |
| Malamute | 1.22 |
| Cocker Spaniel | 1.17 |
| Mixed | 1.05 |
* Odds of having serum thyroid hormone autoantibodies (THAA) among breeds with an increased risk of having THAA, compared with dogs of all other breeds.
From Nachreiner RF, et al: Prevalence of serum thyroid hormone autoantibodies in dogs with clinical signs of hypothyroidism. JAVMA 220:466, 2002.
LYMPHOCYTIC THYROIDITIS AND IMMUNOENDOCRINOPATHY SYNDROMES (POLYGLANDULAR AUTOIMMUNE SYNDROMES).
Because autoimmune mechanisms play an important role in the pathogenesis of lymphocytic thyroiditis, it is not surprising that lymphocytic thyroiditis may occur with other immune-mediated endocrine deficiency syndromes. In humans, two polyglandular autoimmune syndromes, type I and type II, predominate (Table 3-4). Polyglandular autoimmune syndrome type II (Schmidt’s syndrome) is the most common of the immunoendocrinopathy syndromes in humans and is usually defined by the occurrence of primary adrenal insufficiency in combination with autoimmune thyroid disease, insulin-dependent diabetes mellitus, or both (Neufeld et al, 1981). Combinations of endocrine deficiency disorders (e.g., hypothyroidism and diabetes mellitus; Addison’s disease and hypothyroidism) have been documented in dogs (Hargis et al, 1981; Haines and Penhale, 1985; Bowen et al, 1986; Ford et al, 1993; Kooistra et al, 1995; Greco, 2000), although occurrence is uncommon (Table 3-5). In a retrospective study of 225 dogs with hypoadrenocorticism, 4% of the dogs also had hypothyroidism, 0.5% had concurrent diabetes mellitus, and one dog had concurrent hypothyroidism, diabetes mellitus, and hypoparathyroidism (Peterson et al, 1996).
TABLE 3-4 COMPONENT DISORDERS OF TYPE I AND TYPE II POLYENDOCRINE AUTOIMMUNE SYNDROMES
| Type I (% Occurrence) | Type II (% Occurrence) |
|---|---|
| Adrenal insufficiency (100%) | Adrenal insufficiency (100%) |
| Hypoparathyroidism (76%) | Autoimmune thyroid disease (69%) |
| Mucocutaneous candidiasis (73%) | Insulin-dependent diabetes mellitus (52%) |
| Gonadal failure (17%) | Gonadal failure (4%) |
| Autoimmune thyroid disease (11%) | Diabetes insipidus (rare) |
| Insulin-dependent diabetes mellitus (4%) | Hypopituitarism (rare) |
| Miscellaneous Disorders | |
| Alopecia (32%) | Vitiligo (5%) |
| Malabsorption (22%) | Pernicious anemia (1%) |
| Pernicious anemia (13%) | Alopecia (1%) |
| Chronic active hepatitis (13%) | Myasthenia gravis |
| Vitiligo (8%) | Collagen vascular disease |
| Celiac disease | |
Data from Neufeld M, et al: Two types of autoimmune Addison’s disease associated with different polyglandular autoimmune (PGA) syndromes. Medicine 60:355, 1981.
TABLE 3-5 NUMBER OF DOGS WITH VARIOUS ENDOCRINE DEFICIENCY SYNDROMES, SINGLY AND IN COMBINATIONS, SEEN AT OUR HOSPITAL BETWEEN 1990 AND 1994
| Endocrine Disorder | Number of Dogs |
|---|---|
| Insulin-dependent diabetes mellitus | 153 |
| Primary hypothyroidism | 124 |
| Primary hypoadrenocorticism | 72 |
| Primary hypoparathyroidism | 13 |
| Diabetes mellitus and hypothyroidism | 13 |
| Diabetes mellitus and hypoadrenocorticism | 9 |
| Hypoadrenocorticism and hypothyroidism | 3 |
| Diabetes mellitus, hypoadrenocorticism, and hypothyroidism | 2 |
| Hypoparathyroidism and other endocrine deficiencies | 0 |
The initial lesion and precipitating events that result in these syndromes are unknown in dogs. Genetic predisposition to polyglandular autoimmune syndromes has been confirmed in humans (Eisenbarth and Jackson, 1992) and may play a role in the dog as well. Type II polyglandular autoimmune syndrome is the most common form in humans and is inherited as an autosomal dominant trait associated with human leukocyte antigens (HLA; Verge, 1998). Lymphocytic and plasmacytic destruction of affected endocrine glands is identified histologically, and circulating organ-specific autoantibodies are commonly present. Environmental factors combined with an HLA-associated genetic predisposition are thought to trigger the destructive process. Both cell-mediated and humoral immunity appear to be involved in the destruction of target tissues. The most consistent abnormality is a functional defect leading to decreased suppressor T cell immunity. Lymphocytic and plasmacytic destruction of endocrine glands (e.g., lymphocytic thyroiditis, lymphocytic insulitis, lymphocytic adrenalitis) and circulating organ-specific autoantibodies (e.g., thyroglobulin autoantibodies, anti-islet cell antibodies, antiadrenal gland antibodies) have been identified in affected dogs (Haines and Penhale, 1985; Kooistra et al, 1995), findings that suggest etiopathogenic mechanisms in dogs similar to those that have been identified in human beings.
IDIOPATHIC ATROPHY.
Idiopathic atrophy of the thyroid gland is characterized microscopically by loss of the thyroid parenchyma, which is replaced by adipose tissue (Fig. 3-5). An inflammatory infiltrate is lacking, even in areas in which small follicles or follicular remnants are present (Gosselin et al, 1981b) and tests for lymphocytic thyroiditis are negative. The parathyroid glands are rarely affected, and variable numbers of parafollicular cells remain.
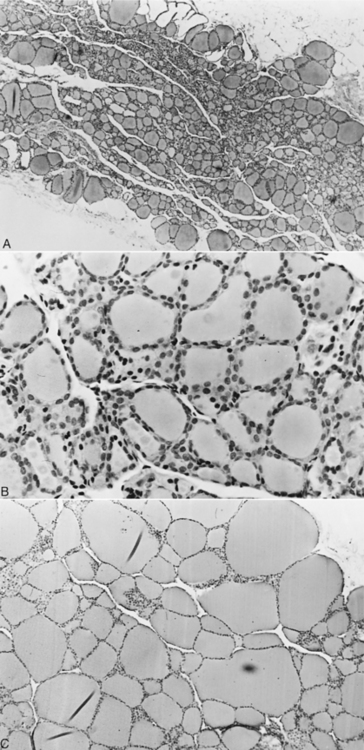
The cause of idiopathic thyroid atrophy is not known. It has been suggested that idiopathic atrophy is a primary degenerative disorder involving individual follicular cells (Gosselin et al, 1981b). Degeneration of follicular cells is seen histologically early in the lesion, with their subsequent exfoliation into the colloid or the interfollicular spaces. Progressive reduction in the size of the follicles and replacement of the degenerating follicles with adipose tissue occur. This atrophy can be distinguished from the atrophy associated with decreased TSH secretion (i.e., secondary hypothyroidism), in which the follicles are lined by low cuboidal epithelial cells with no indication of degeneration.
Atrophy of the thyroid gland may also represent an end stage of lymphocytic thyroiditis. Evaluation of the morphologic changes involved in lymphocytic thyroiditis in a colony of related Borzoi dogs revealed initial degenerative thyroidal parenchymal changes, which progressed to progressively worsening inflammation, subsequent fibrosis, and an end stage of thyroid gland destruction, which manifested itself as an entity histologically similar to idiopathic follicular atrophy (Conaway et al, 1985b). However, residual inflammation was still evident histologically at the end stage of lymphocytic thyroiditis. In a recent study, the mean age at the time of diagnosis of hypothyroidism was older in dogs with suspected idiopathic atrophy, compared with dogs diagnosed with lymphocytic thyroiditis; a finding that supports the theory that idiopathic atrophy may be an end stage of lymphocytic thyroiditis (Graham et al, 2001a). Results for serum thyroglobulin and thyroid hormone autoantibody tests also progress from positive to negative with time in dogs with lymphocytic thyroiditis, suggesting that the inciting antigens for lymphocytic thyroiditis disappear with time. Although idiopathic atrophy may represent an end-stage form of autoimmune lymphocytic thyroiditis, the inability to demonstrate an inflammatory cell infiltrate, even when follicles are still present, on histopathologic examination of the thyroid gland in dogs diagnosed with idiopathic atrophy suggests that there may be more than one etiology for thyroid atrophy in the dog. Unlike for lymphocytic thyroiditis, there are no blood tests currently available that establish the diagnosis of idiopathic atrophy. Hence the diagnosis tends to be one of exclusion; that is, if the tests for lymphocytic thyroiditis are negative, the dog must have idiopathic atrophy. Until histologic evaluation of several thyroid gland biopsies obtained from the same dog over time has been done in a group of dogs in the early stages of naturally acquired lymphocytic thyroiditis, the relationship between lymphocytic thyroiditis and idiopathic atrophy of the thyroid gland will remain conjectural.
FOLLICULAR CELL HYPERPLASIA.
Histologic evaluation of the thyroid gland in some dogs with hypothyroidism reveals small thyroid follicles that contain minimal amounts of colloid. Follicular cell hyperplasia is present, however. No appreciable inflammation is seen within the gland, and circulating antithyroglobulin antibody has been undetectable in a few dogs studied. The cause of these pathologic changes is not known, although the histologic changes resemble those found with iodine deficiency or in juvenile dogs and cats with defects in thyroid hormone biosynthesis (Chastain et al, 1983; Arnold et al, 1984). Iodine deficiency is extremely unlikely because these dogs have been fed nutritionally balanced, commercial dog foods. One possible explanation is impaired function of the follicular cells (e.g., dyshormonogenesis), resulting in a thyroid-deficient state. Follicular cell hyperplasia may develop secondary to the stimulatory effects of increased TSH secretion in response to thyroid hormone deficiency, assuming appropriate binding of TSH to its receptor occurs. It is interesting to note that ingestion of diets containing an excessive amount of iodine can cause significant impairment of thyroid function and hypothyroidism (Castillo et al, 2001). Excessive iodine intake inhibits iodide uptake and organification and thyroid hormone secretion by thyroid follicular cells, resulting in a compensatory increase in circulating TSH concentrations (Pisarev and Gartner, 2000).
NEOPLASTIC DESTRUCTION.
In most dogs, thyroid tumors are hormonally inactive and do not secrete excessive amounts of thyroid hormone. As a result, clinical signs of hyperthyroidism are uncommon. In our hospital, 55% to 60% of dogs with thyroid neoplasia have normal serum thyroid hormone concentrations, 30% to 35% have low serum thyroid hormone concentrations and clinical signs supportive of hypothyroidism, and approximately 10% have increased serum thyroid hormone concentrations and signs supportive of hyperthyroidism (see Chapter 5).
Although most canine thyroid tumors have been considered “nonfunctioning,” they may secrete inactive or altered forms of thyroid hormone. The altered hormone may not cause thyrotoxicosis and may not be detected by standard radioimmunoassay techniques. These products, however, may be capable of suppressing TSH secretion, in which case atrophy of the remaining normal thyroid tissue would result. This process was thought to have occurred in one dog described as having hypothyroidism and a follicular thyroid carcinoma involving only one lobe of the thyroid gland (Branam et al, 1982).
MISCELLANEOUS CAUSES.
Primary hypothyroidism may result from the ingestion of toxic agents or antithyroid medications (e.g., propylthiouracil, methimazole) or following surgical removal of a thyroid tumor. Accessory thyroid tissue is common in dogs and may be found from the base of the tongue to the base of the heart. Therefore surgical removal of the thyroid gland rarely results in permanent hypothyroidism. Use of radioactive iodine (iodine-131) to treat hyperthyroidism may result in total ablation of the thyroid, especially if both thyroid lobes are affected and excessive doses are used (Meric et al, 1986).
SUMMARY.
Although there are several potential causes (see Table 3-1), lymphocytic thyroiditis and idiopathic atrophy account for most of the clinical cases of primary hypothyroidism diagnosed in dogs. Both cause progressive loss of thyroid function as a result of either immune-mediated destruction (lymphocytic thyroiditis) or degeneration (idiopathic atrophy) of the thyroid. The result is a deficiency in thyroid hormone synthesis and secretion and development of clinical signs of hypothyroidism.
Secondary Hypothyroidism
Potential causes of secondary hypothyroidism include congenital malformations of the pituitary gland, pituitary destruction, and pituitary suppression. In the dog, secondary hypothyroidism caused by naturally acquired defects in pituitary thyrotroph function or destruction of pituitary thyrotrophs (e.g., pituitary neoplasia) is uncommon. In contrast, suppression of pituitary thyrotroph function by hormones or drugs (e.g., glucocorticoids, naturally occurring hyperadrenocorticism) is quite common. Serum TSH concentrations should be decreased or undetectable with secondary hypothyroidism. Unfortunately, current assays used to measure endogenous TSH in dogs are unable to differentiate between normal and decreased concentrations (see page 118), making confirmation of secondary hypothyroidism difficult. Identification of an increased serum TSH concentration is consistent with primary hypothyroidism, and although one would expect the serum TSH concentration to be undetectable in a dog with secondary hypothyroidism, such a finding does not confirm the diagnosis.
PITUITARY MALFORMATION.
Congenital abnormalities involving the pituitary gland have been recognized in many breeds but are reported most commonly in German Shepherd dogs. Cystic Rathke’s pouch or hypoplasia of the anterior pituitary affecting the thyrotropic cells results in impaired secretion of TSH (Eigenmann, 1981; Hamann et al, 1999; Kooistra et al, 2000a). Because of the involvement of other anterior pituitary hormones, most notably growth hormone (GH), congenital defects affecting the anterior pituitary usually result in the development of dwarfism (see Chapter 2). If TSH but not GH secretion were affected, cretinism would develop (see page 104).
PITUITARY THYROTROPH SUPPRESSION.
Secondary hypothyroidism may develop following suppression of thyrotroph function by concurrent illness, drugs, hormones, or malnutrition (see Factors Affecting Thyroid Gland Function Tests, page 126). These are the most common causes of secondary hypothyroidism in the dog. From a clinical standpoint, perhaps the most important is the suppressive effect of glucocorticoids, either from exogenous administration or from naturally acquired hyperadrenocorticism. In a study of 102 dogs with naturally acquired hyperadrenocorticism, randomly obtained basal serum T4 and/or serum T3 levels were decreased in 68% of the dogs (Ferguson and Peterson, 1986). The thyroid is usually subnormally responsive to TSH administration (see Chapter 6). Unlike the other causes, secondary hypothyroidism induced by suppression of thyrotroph function is potentially and usually reversible. Thyroid hormone supplementation is not required if the cause for the suppression can be identified and treated.
MISCELLANEOUS CAUSES.
In humans, secondary hypothyroidism may also develop following production of a defective TSH molecule or impaired interaction between TSH and its receptor on follicular epithelial cells. These causes have not yet been reported in the dog. Secondary hypothyroidism can follow radiation therapy for expansile adrenocorticotrophic hormone (ACTH)–secreting pituitary tumors. Secondary hypothyroidism can also be caused by hypophysectomy (Lantz et al, 1988; Meij et al, 1998).
Tertiary Hypothyroidism
Tertiary hypothyroidism is defined as a deficiency in the secretion of thyrotropin-releasing hormone (TRH) by peptidergic neurons in the supraoptic and paraventricular nuclei of the hypothalamus. Lack of TRH secretion should cause a deficiency in TSH secretion and secondary follicular atrophy in the thyroid gland. Histologically, the thyroid gland would then resemble glands seen in dogs with secondary hypothyroidism. In humans, impaired secretion of TRH by the hypothalamus may result from a congenital defect, acquired destruction secondary to a mass lesion or hemorrhage, a defective TRH molecule, or defective TRH-thyrotroph receptor interaction (Larsen et al, 1998). Neurologic signs and additional pituitary dysfunction may be present, depending on the cause. In theory, differentiation between secondary and tertiary hypothyroidism would rely on changes in the serum TSH concentration after administration of TRH (see page 120). Tertiary hypothyroidism has not been reported in dogs and can be assumed to be rare.
Congenital Defects
The incidence of congenital hypothyroidism in the dog is not known. In a survey of 2642 dogs with hypothyroidism, 3.6% were less than 1 year of age (Milne and Hayes, 1981). It can be assumed that a percentage of these animals had congenital defects of the thyroid gland. Unfortunately, congenital hypothyroidism probably results in early death of most affected puppies, and the cause of death is inadvertently lumped into the broad diagnosis of “fading puppy syndrome.” Thus most puppies with congenital hypothyroidism are not diagnosed or documented.
Congenital defects are divided into four categories in humans: thyroid dysgenesis, dyshormonogenesis, circulating thyroid hormone transport abnormalities, and metabolic defects following ingestion of goitrogens. Mutations in the TSH molecule that cause it to be ineffective as a thyroid stimulator or a defect in the TSH receptor that impairs the ability of the thyroid cell to respond to TSH have also been described in humans (Hayashizaki et al, 1990; Sunthornthepvarakul et al, 1994). In humans, the majority of congenital thyroid defects are due to thyroid dysgenesis (i.e., aplasia, hypoplasia, or ectasia; Larsen et al, 1998). Of the remaining categories, an inherited inability to organify iodide is the most common cause of congenital hypothyroidism in human infants. Only a few reports of canine congenital hypothyroidism have appeared in the literature. Documented causes of congenital primary hypothyroidism in the dog include deficient dietary iodine intake, dyshormonogenesis (i.e., iodine organification defect), and thyroid dysgenesis (Chastain et al, 1983; Greco et al, 1985). Secondary hypothyroidism resulting from an apparent deficiency of TSH has also been reported in a family of Giant Schnauzers (Greco et al, 1991) and a Boxer dog (Mooney and Anderson, 1993). Pedigree analysis suggested an autosomal recessive mode of inheritance in the family of Giant Schnauzers. Pituitary dwarfs with combined anterior pituitary hormone deficiencies usually lack TSH in addition to GH and prolactin (Hamann et al, 1999; Kooistra et al, 2000a; see Chapter 2). Lack of TSH secretion may contribute to abnormal body maturation and growth in pituitary dwarfs.
The development of an enlarged thyroid gland (i.e., goiter) depends on the etiology. If the hypothalamic-pituitary-thyroid gland axis is intact, appropriate binding of TSH occurs with its receptor, and the block in thyroid hormone production is within the thyroid gland itself (e.g., as occurs with an iodine organification defect), goiter will develop in response to increased serum TSH concentration. Treatment with levothyroxine sodium will cause a decrease in the serum TSH concentration, providing evidence for normal pituitary thyrotrophs. If the hypothalamic-pituitary-thyroid gland axis is not intact (e.g., as occurs with pituitary TSH deficiency), goiter will not develop. Serum TSH concentration is decreased, although current TSH assays used in dogs cannot detect low TSH concentrations (see page 118). If defects exist in the binding of TSH to its receptor, serum TSH concentration will be increased and goiter absent. TSH binding defects have not been documented in dogs.
An inability to convert T4 to T3 by peripheral tissues, presumably as a result of a deficiency in 5’-monodeiodinase, is often proposed to explain low serum T3 but normal serum T4 concentrations in some dogs. Unfortunately, conversion defects have not been documented in humans or dogs and are probably rare, if they exist at all. Seemingly, a deficiency in 5’-monodeiodinase would be incompatible with life and should cause fetal resorption, fetal abortion, or neonatal death (i.e., “fading puppy syndrome”). Low serum T3 concentrations in conjunction with normal T4 levels are quite common in euthyroid dogs because of normal fluctuations in serum T3 concentration, concurrent illness, and drug therapy (see page 115). Nondetectable serum T3 concentrations can also occur in dogs with lymphocytic thyroiditis and T3 autoantibodies in the circulation. Depending on the type of assay used to measure T3 concentrations, T3 autoantibodies may interfere with the assay and cause spuriously low results (see page 125).
CLINICAL FEATURES OF PRIMARY HYPOTHYROIDISM IN THE ADULT DOG
Signalment
No single diagnostic test confirms the diagnosis of hypothyroidism in the dog. Therefore reports containing data regarding breed incidence and genetics for this disorder should always be examined critically, especially in terms of how the diagnosis of hypothyroidism was made. Evaluation for serum thyroglobulin and thyroid hormone autoantibodies provides insight into breeds that have an increased incidence of lymphocytic thyroiditis (Table 3-3) and may indicate a genetic role for the disorder in some of these breeds (Nachreiner et al, 2002). Local breed popularity can also influence a veterinarian’s perception of which breeds are predisposed to developing hypothyroidism, a perception that may or may not be accurate. In our hospital, hypothyroidism is most commonly diagnosed in Doberman Pinschers, Golden Retrievers, Labrador Retrievers, and Cocker Spaniels (Table 3-6). Clinical signs usually develop during middle age (i.e., 2 to 6 years). Of 3206 dogs diagnosed with hypothyroidism, 32% were between 4 and 6 years of age, and 22% each were between 2 and 3 years and 7 and 9 years of age (Milne and Hayes, 1981). Age of onset of symptomatic hypothyroidism may vary between breeds, presumably as a result of the underlying etiology and rate of progression of thyroid pathology. Individual variability also exists within a breed. In general, breeds at increased risk tend to develop clinical signs at an earlier age than other breeds (Milne and Hayes, 1981; Nesbitt et al, 1980; Muller et al, 1983). There is no apparent gender predisposition.
TABLE 3-6 BREED DISTRIBUTION OF 130 DOGS WITH PRIMARY HYPOTHYROIDISM
| Breed | Number (%) of Dogs |
|---|---|
| Golden Retriever | 24 (18%) |
| Doberman Pinscher | 22 (17%) |
| Labrador Retriever | 8 (6%) |
| Cocker Spaniel | 7 (5%) |
| German Shepard dog | 7 (5%) |
| Mixed Breed | 7 (5% |
| Dachshund | 5 (4%) |
| Poodle | 4 (3%) |
| Rottweiler | 4 (3%) |
| Spaniels (Springer, King Charles) | 4 (3%) |
| Akita | 3 (2%) |
| Boxer | 3 (2%) |
| Terriers (Fox, Scottish, Westie) | 3 (2%) |
| Beagle | 2 (2%) |
| Chesapeake Bay Retriever | 2 (2%) |
| Chow Chow | 2 (2%) |
| Maltese | 2 (2%) |
| Mastiff | 2 (2%) |
| Old English Sheepdog | 2 (2%) |
| Samoyed | 2 (2%) |
| Shetland Sheepdog | 2 (2%) |
| 13 breeds | < 1% each |
Clinical Signs
Thyroid hormone is needed for the normal cellular metabolic functions of the body. A deficiency in circulating thyroid hormone affects the metabolic function of almost all organ systems. As a result, clinical signs are quite variable and depend in part on the age of the dog at the time a deficiency in thyroid hormone develops (Table 3-7). Clinical signs may also differ between breeds. For example, different breeds appear to have markedly different hair cycles and follicular morphology, which may influence the clinical and histologic features of the disease (Credille et al, 2001). Similarly, dermatologic signs may predominate in some breeds, whereas neuromuscular signs may predominate in other breeds. Destruction of the thyroid gland tends to be a slow process, often taking more than a year from the time tests for lymphocytic thyroiditis are positive until tests of thyroid gland function become abnormal. Hence the onset of clinical signs is often gradual and subtle, and this can hamper the clinician’s ability to establish the diagnosis. Important information regarding the existence of some clinical signs (e.g., metabolic signs) may not be evident from the history simply because the clinical signs have been slow to develop, the owners have unconsciously adapted to the changes in their dog, and they do not recognize the problem. Only after the dog returns to normal following initiation of thyroid hormone supplementation does the owner have a reference point and can recognize that a problem existed.
TABLE 3-7 CLINICAL MANIFESTATIONS OF HYPOTHYROIDISM IN THE ADULT DOG
< div class='tao-gold-member'> Only gold members can continue reading. Log In or Register to continue
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|