Chapter 16 HYPERCALCEMIA AND PRIMARY HYPERPARATHYROIDISM
HYPERCALCEMIA
INTRODUCTION
Calcium is required for numerous vital intracellular and extracellular functions. For example, calcium is necessary for various enzymatic reactions, transport of substances across membranes and membrane stability, blood coagulation, nerve conduction, neuromuscular transmission, muscle contraction, smooth muscle tone, hormone secretion, bone formation, control of hepatic glycogen metabolism, and cell growth and division (Rosol et al, 2000). Calcium serves as an almost universal ionic messenger, conveying signals received at cell surfaces into cells (Rasmussen, 1989). In the extracellular fluid, calcium helps regulate the cellular function of various organs, including the parathyroid glands, kidneys, and thyroid C cells (Brown et al, 1995).
The concentration of serum ionized calcium (physiologically “active” calcium) is normally maintained within narrow limits by the action of parathyroid hormone (PTH) on bone resorption, renal calcium excretion, and metabolism of vitamin D (Table 16-1). PTH (primarily an 84–amino acid, single-chain polypeptide) is synthesized, stored, and secreted by chief cells in the four parathyroid glands. Secretion is regulated, in turn, by the serum calcium concentration: high concentrations inhibit secretion of PTH, and low concentrations stimulate it (Aurbach et al, 1985a; Brown et al, 1999). The amino acid sequence of PTH is known for humans, dogs, cows, pigs, rats, chickens, and cats. Based on immunologic reactivities, most mammals appear to have similar amino-terminal portions of the molecule (Toribio et al, 2002). It is recognized that the amino terminal of PTH binds to cell membrane receptors. The carboxyl terminus is thought to serve only as a guide for PTH through the cellular secretory pathway (Orloff and Stewart, 1995). A synthetic 1-34 amino-terminal fragment is biologically active, but relatively minor modifications at the amino terminal (especially at the first two residues) can completely abolish its biologic activity (Marx, 2000). The effect of PTH on mineral metabolism is initiated by binding of PTH to type 1 receptors in target tissues, which results in regulation of large calcium fluxes across bone, kidneys, and intestines (Fig. 16-1).
TABLE 16-1 NORMAL SERUM CONCENTRATIONS*
| Dog | Cat | |
|---|---|---|
| Total Calcium | ||
| (mg/dl) | 9.0–11.7 | 8.0–10.5 |
| (mmol/L) | 2.2–3.9 | 2.0–2.6 |
| Ionized Calcium | ||
| (mg/dl) | 4.6–5.6 | 4.5–5.5 |
| (mmol/L) | 1.12–1.42 | 1.1–1.4 |
| Parathyroid Hormone | ||
| (pmol/L) | 2–13 | 0–4 |
| Parathyroid Hormone- | ||
| Related Protein (PTH-P) | ||
| (pmol/L) | <2 | <2 |
| 1,25-Dihydroxyvitamin | ||
| D (Calcitriol) | ||
| (pg/ml) | ||
| Adults | 20–50* | 20–40* |
| 10–12 wk old | 60–120* | 20–80* |

Parathyroid hormone–related peptide (PTHrP) is a distant homologue of PTH and is not considered a true hormone. It is produced widely in the body and has numerous actions in the normal fetus and adult. However, this substance is best recognized as having a central role in the pathogenesis of humoral hypercalcemia of malignancy and is used as a “tumor marker.” Since its discovery in the early 1980s, assay of PTHrP has been used in the evaluation of hypercalcemic patients in whom cancer is a possibility (Philbrick et al, 1996).
ETIOLOGY
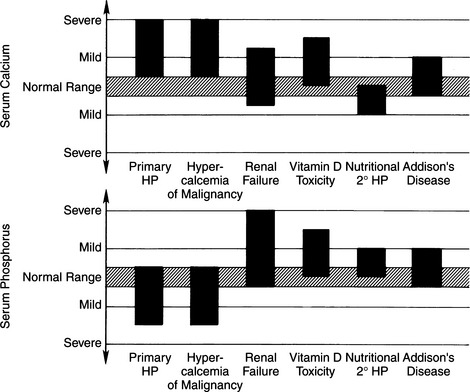
The essential disorder in primary hyperparathyroidism (PHPTH) is the excessive synthesis and secretion of PTH by abnormal, autonomously functioning parathyroid “chief” cells. The cause of this hormonal excess is usually a solitary adenoma, but adenomatous hyperplasia of one or more parathyroid glands and parathyroid carcinomas have been identified in both dogs and cats. In contrast, other forms of hyperparathyroidism (e.g., renal secondary or nutritional secondary hyperparathyroidism) are usually the result of alterations in calcium and phosphorus homeostasis caused by nonendocrine disturbances. Such disturbances indirectly affect the parathyroid glands, causing diffuse parathyroid gland hyperplasia (see Differential Diagnosis of Hypercalcemia, page 669). Depending on the underlying cause, the serum calcium concentration in secondary disorders may range from low to normal to increased. Primary hyperparathyroidism is virtually always associated with hypercalcemia, as reflected by increases in circulating concentrations of the total or ionized calcium concentration or both.
PATHOPHYSIOLOGY
Severe Hypercalcemia, Bone Resorption, and the Kidneys
BONE.
The process leading to severe hypercalcemia is initiated by accelerated bone resorption caused by the activation of osteoclasts in the skeleton (Attie, 1989). The activity of these cells, caused by PTH or PTHrP, is the foundation for virtually all cases of marked hypercalcemia. Excessive absorption of calcium from the gastrointestinal tract is not usually an important cause of hypercalcemia, although it is a contributor to the hypercalcemia resulting from vitamin D toxicosis. Hypercalcemia develops when the entry of calcium into the extracellular fluid (regardless of the source) overwhelms the mechanisms that maintain normocalcemia. One of these mechanisms is suppressed secretion of PTH, a process obviously negated when the cause of the hypercalcemia is the autonomous secretion of PTH. In cancer-associated hypercalcemia, the secretion of PTH is suppressed, but the humoral factor that activates osteoclasts is the autonomous secretion of PTHrP. PTHrP, as previously discussed, has biologic activity quite similar to that of PTH (Sleeboom and Bijvoet, 1982; Harinck et al, 1987). In the setting of accelerated bone resorption, the kidneys become the principal defense against hypercalcemia (Bilezikian, 1992a; Hruska and Teitelbaum, 1995).
Associated with accelerated osteocytic and osteoclastic bone resorption, mineral is removed and replaced by immature fibrous connective tissue. The bone lesion of fibrous osteodystrophy in afflicted dogs is reported to be generalized throughout the skeleton but accentuated in certain areas, such as the cancellous bone of the skull (Capen and Martin, 1983). Iliac crest bone biopsies from virtually all humans with primary hyperparathyroidism reveal histomorphometric evidence of the effects of excess PTH (Arnaud and Kolb, 1991; Hruska and Teitelbaum, 1995). These findings may include increased bone resorption surfaces, increased numbers of osteoclasts, osteocytic osteolysis, and marrow fibrosis. Bone histomorphometric findings in dogs with primary hyperparathyroidism were similar to those reported in humans (i.e., parallel increases in osteoblastic and osteoclastic activity). The increased rate of bone formation in these dogs was reflected in an increased rate of mineral apposition and a greater extent of tetracycline-labeled bone-forming surface (Meuten et al, 1983a; Weir et al, 1986).
KIDNEYS.
When renal and endocrine functions are normal, any tendency for a rise in serum calcium is attenuated by increased urinary excretion of calcium. This process is inhibited by PTH excess, the foundation of primary hyperparathyroidism. Severe hypercalcemia (defined as a serum calcium concentration >14 mg/dl in humans and, arbitrarily, >15 mg/dl in dogs) is the result of humoral factors (PTH or PTHrP) that induce osteoclast-mediated bone resorption and stimulate intestinal absorption of calcium and renal tubular resorption of calcium (Mundy, 1988; Hruska and Teitelbaum, 1995). This impairs the kidneys’ ability to excrete the increased filtered load of calcium. Thus animals with an excess of PTH lack the first lines of defense against hypercalcemia.
Depletion of the extracellular fluid volume and a reduced glomerular filtration rate (GFR) as a result of dehydration may further increase the serum calcium concentration (Bilezikian, 1991). Hypercalcemia alters the GFR by causing (1) volume contraction of the extracellular fluid space secondary to fluid losses; (2) alterations in glomerular capillary permeability; and/or (3) sustained vasoconstriction that may result in ischemic injury to the kidneys, directly potentiating the toxic effects of hypercalcemia on renal tubular cells. Furthermore, mineralization of tissues may begin in the basement membrane of the ascending loop of Henle, the distal convoluted renal tubule, and the collecting ducts. This damages the basement membrane and impairs renal tubular repair. At the organelle level, mineralization of mitochondria occurs in renal tubular epithelial cells, potentially leading to disrupted cellular function, death, and sloughing of mineralized cell debris into renal tubular lumina. Casts composed of renal tubular cells may contribute to obstruction of urine flow along affected nephrons.
Nephrocalcinosis may be mild and reversible or severe and progressive, leading to continued renal damage and uremia. Severe mineralization of the kidneys is characteristically located subjacent to the corticomedullary junction and has the appearance of a white gritty band (Meuten, 1984). Histologic findings include interstitial infiltration of mononuclear cells, varying degrees of renal tubular atrophy, calcification, necrosis, and sloughing of the epithelium. In humans, renal failure may initially result from prerenal factors, but if hypercalcemia persists, the failure may be the result of acquired intrinsic renal lesions. It is extremely important to point out that this series of physiologic events described in humans seems to be much less worrisome in dogs with PHPTH. Almost all dogs with PHPTH have normal blood urea nitrogen (BUN) and serum creatinine concentrations. Furthermore, as we have monitored a considerable number of untreated dogs for periods longer than 12 to 18 months, it is safe to say that we have observed no evidence of progressive renal damage. To the contrary, the kidneys in dogs appear to be “protected” by PHPTH. As has been noticed in untreated humans, virtually no dramatic changes in serum biochemical profiles (aside from increasing calcium and decreasing phosphate concentrations) are noted over time (Silverberg et al, 1999).
Uroliths and Urinary Tract Infections
Reports on dogs with primary hyperparathyroidism (PHPTH) have also included a percentage of dogs (˜30%) with urinary calculi (Berger and Feldman, 1987; Klausner et al, 1987). Hypercalciuria results from increased glomerular filtration of calcium, which we have always presumed to predispose these animals to urolithiasis. However, other hypercalcemic conditions have not been associated with urolithiasis. This discrepancy may be explained by the chronic nature of the hypercalcemia in dogs with PHPTH, because many tend to be asymptomatic or to have only mild signs, as opposed to the short-term nature of hypercalcemia in dogs with neoplasia, toxin exposure, and other such conditions. In these other conditions, dogs tend to develop obvious or severe clinical signs quickly and, therefore, do not have chronic hypercalcemia. Additional factors may play a role in urolith formation or urinary solute saturation, including pH, ionic strength, and crystal aggregation inhibitors. Calcium phosphate, for example, is markedly less soluble in alkaline urine than in acidic urine.
Vitamin D Excesses and Deficiencies
An adaptive mechanism to increases in the serum calcium concentration is a decrease in the circulating vitamin D concentration, assuming that the hypercalcemia is not a result of a primary renal or endocrine disorder. Decreases in the vitamin D concentration should result in reduced calcium absorption from the intestine. By contrast, the response to mild decreases in the serum calcium concentration is secretion of both PTH and vitamin D. PTH-stimulated bone resorption and vitamin D–stimulated intestinal absorption of both calcium and phosphorus then stimulate a rise in the serum calcium and phosphate concentrations (see Fig. 16-1). The renal tubular effects of PTH aid mineral homeostasis by stimulating synthesis and secretion of 1,25[OH]2D3 (vitamin D). PTH also functions at the renal tubular level to increase phosphate excretion into the urine. This is important for preventing hyperphosphatemia after phosphate is removed from bone with calcium and/or absorbed from the intestine with calcium.
Vitamin D deficiency, after chronic PTH-stimulated use of vitamin D stores, is a possible natural adaptive mechanism for coping with primary hyperparathyroidism. This process seems unlikely in dogs with PHPTH because humans with vitamin D deficiency caused by chronic hyperparathyroidism typically have severe osteomalacia, a problem not yet reported in animals (Meuten et al, 1983a; Weir et al, 1986). Furthermore, in a study that included four dogs with primary hyperparathyroidism, three had calcitriol concentrations that were mildly increased or in the high normal range (Rosol et al, 1992a). If the results in these three dogs accurately reflect the physiology of most dogs with PHPTH, osteomalacia would be uncommon, and the previously discussed physiologic triad is better understood.
Renal Effects of Hypercalcemia and Soft Tissue Mineralization
Several natural adaptive mechanisms are initiated in an attempt to control and possibly correct the hypercalcemia associated with primary hyperparathyroidism. One mechanism is the previously described hypercalciuria. Another occurs once the renal tubular transport of calcium is overwhelmed. When the solubility product of the serum calcium (Ca++) and phosphate (PO4) concentrations exceed 60 to 80 mg/dl, deposition of calcium in soft tissue ensues (Meuten et al, 1981). The result of this trade-off to lower the serum calcium concentration is the development of progressive organ dysfunction, especially in the kidneys as a result of nephrocalcinosis. Soft tissue calcification may occur in a variety of other organs or tissues (or both), but none is quite as worrisome as that which occurs in the kidneys.
As discussed previously, azotemia occurs commonly in dogs with hypercalcemia of malignancy, hypoadrenocorticism, chronic renal failure (CRF), and hypervitaminosis D (Kruger et al, 1996). Hypercalcemia can contribute to azotemia with any combination of the following: prerenal reduction in extracellular fluid volume (anorexia, hypodipsia, vomiting, and polyuria); renal vasoconstriction; decreased permeability coefficient of the glomerulus (Kf); acute tubular necrosis from the ischemic and toxic effects of hypercalcemia; and CRF caused by nephron loss, nephrocalcinosis, tubulointerstitial inflammation, and interstitial fibrosis (Rosol et al, 2000).
The frequency of azotemia was higher in dogs with malignancy (71%) than in those with PHPTH (11%) (Kruger et al, 1996). It is our impression, however, that CRF is almost never caused by PHPTH in dogs. The incidence of CRF in our series of dogs with PHPTH is approximately 4%. It is likely that a similar incidence of CRF would be identified in any population of dogs in which the average age is 10 to 11 years. The most common cause of CRF in hypercalcemic dogs would be related to a calcium × phosphorus product above 60 to 80 mg/dl. Such increases in this product would be extremely unusual in PHPTH because of the low-normal or low serum phosphorus concentrations caused by PTH-induced excesses in vitamin D. Therefore, although a typical normal dog has a total serum calcium (Ca) concentration of about 10.5 mg/dl, a serum phosphate of about 4.5 mg/dl, and a product of 47, the typical dog with PHPTH has a total serum Ca concentration of about 15 mg/dl, a serum phosphate of about 3.0 mg/dl, and a product of 45. This is in contrast to a dog with CRF or vitamin D toxicosis, with a total serum Ca concentration of about 10.5 mg/dl (or higher), a serum phosphate of about 10.0 mg/dl (or higher), and a product in excess of 100.
Rapid Metabolism of PTH
Stimulation, via hypercalcemia, of rapid tissue degradation of the biologically active forms of PTH by parathyroid and nonparathyroid cells may represent a physiologic mechanism to curtail worsening hypercalcemia in PHPTH (Arnaud and Kolb, 1991). Plasma calcium may regulate PTH secretion and also determine the circulating quantities of biologically active PTH molecules versus inactive hormone fragments. This phenomenon may be more theoretical than practical because the disappearance rate of PTH from serum after the removal of a parathyroid tumor is no different from that of dogs with nonparathyroid disease in which the parathyroids are surgically removed.
Calcitonin
Calcitonin, a 32–amino acid polypeptide hormone produced by parafollicular or “C cells” located in the thyroid, is secreted in response to increases in serum calcium (Mol et al, 1991). This hormone has the primary responsibility of limiting postprandial hypercalcemia in normal mammals. Calcitonin decreases bone resorption by reducing the size of osteoclast brush borders, the number of osteoclasts, and osteoclast motility. Calcitonin does not affect the kidney or intestine. It may, however, influence the satiety center, decreasing appetite. The role of calcitonin in dogs with hypercalcemia secondary to PHPTH is not known. Increased calcitonin secretion in response to hypercalcemia seems a reasonable physiologic response. Parafollicular cells are markedly hyperplastic and appear as small white foci in the thyroid gland of dogs with primary hyperparathyroidism (Capen and Martin, 1983). Hyperplastic parafollicular cells may displace colloid-containing follicles lined by thyroid follicular cells, implying the presence of an adaptive response by the parafollicular cells. Studies in humans suggest that this adaptive mechanism is inconsistent (Arnaud and Kolb, 1991).
ASSAYS OF SERUM PARATHYROID HORMONE
PTH Assay Systems
Development of the “two-site” PTH assay system currently used by most human and veterinary laboratories depends on the production of two different polyclonal antibodies. The two-site immunoradiometric (IRMA) system for intact human PTH (Allegro Intact PTH; Nichols Institute, San Juan Capistrano, Calif.) has been demonstrated to be valid for measurement of dog and cat PTH (Torrance and Nachreiner, 1989a; Flanders and Reimers, 1991; Barber et al, 1993). One antibody binds only the midregion and C-terminal 39-84 amino acids of human PTH. The other antibody binds only the N-terminal 1-34 amino acids of human PTH. Samples being assayed are incubated simultaneously with both antibodies, with a “sandwich” created composed of the 1-34 antibody plus the patient’s intact hormone plus the 39-84 antibody. Only these “sandwiches” are detected by the assay system, eliminating any interference by midregion or C-terminal fragments, even if present in large concentrations. Two-site immunochemiluminometric assays for intact PTH in humans use antibodies similar to those in the intact PTH IRMA assays and have the same range of applicability to animal sera (Michelangeli et al, 1997).
Clinical Usefulness
BACKGROUND.
Experience with the new PTH two-site assays has been extremely positive. Samples obtained for this assay should be stored and shipped frozen to prevent degradation of intact PTH. Stability is best achieved in plasma collected with ethylenediamine tetra-acetic acid (EDTA), but serum is adequate if stored frozen after separation from red blood cells. Normal values for the serum PTH concentration are 2 to 13 pmol/L in dogs and 0 to 4 pmol/L in cats. Samples should be shipped to the appropriate laboratory with two or three frozen gel packs (Torrance and Nachreiner, 1989a and 1989b).
As with most hormone assays, evaluation of the serum hormone concentration “out of context” is not as consistently informative as evaluation in the proper context. With this in mind, the serum PTH concentration must be evaluated relative to the total and, ideally, ionized serum calcium concentrations. Decreases in the serum calcium concentration are normally associated with increases in the serum PTH concentration. Increases in the serum calcium concentration are normally associated with decreases in the PTH concentration (Fig. 16-2). As with any laboratory value, a single PTH result may not provide correct information. Therefore practitioners are encouraged to complete a thorough evaluation of hypercalcemic dogs or cats. If test results do not appear logical, some tests may need to be repeated, including measurement of serum hormone concentrations, which may fluctuate.

ASSAYS OF IONIZED SERUM CALCIUM CONCENTRATIONS
The biologically active form of calcium in the circulation is the ionized fraction. Measurement of the serum ionized calcium concentration has become widely available and cost-effective (normal or “reference” concentrations for our laboratory are 1.12 to 1.42 mmol/L; see Table 16-1). Although samples can be obtained routinely rather than according to the previously described anaerobic requirements, anaerobically obtained, handled, and stored serum is stable for a longer time and therefore preferred (Schenck et al, 1995). Anaerobic collection ensures that no increase in pH occurs as a result of loss of carbon dioxide (CO2). An acidic pH favors dissociation of calcium from protein and increases the amount of ionized calcium in the sample. An alkaline pH occurs with loss of CO2 and favors calcium binding to protein, thus reducing the amount of ionized calcium. Mixing of serum and air increases the pH, decreasing the ionized calcium concentration. Ionized calcium and pH are more stable in serum than in whole or heparinized blood. The analysis of serum eliminates potential interference by heparin and allows longer storage periods. Serum may be stored for subsequent ionized calcium analysis as long as it has been handled anaerobically. Silicone separator tubes should not be used. Measurement of ionized calcium in canine serum was stable after storage at 23°C for 72 hours and for 7 days at 4°C (Schenck et al, 1995).
The use of automated equipment with calcium ion–selective electrodes has resulted in easy and accurate measurement that minimizes interference caused by magnesium, potassium, protein, hemolysis, and other factors (Gouget et al, 1988). However, differences among analyzers exist, and it is recommended that each laboratory establish reference ranges for the analyzer they use (Hristova et al, 1995). Some instruments mathematically manipulate the ionized calcium concentration and actual pH value of the sample and yield an “adjusted” value. Formulas have also been developed to “adjust” results for samples exposed to air. These formulas have not been validated in animals, and their use is not recommended (Rosol et al, 2000).
The addition of this diagnostic tool can be valuable in the evaluation of hypercalcemia and hypocalcemia as well as renal insufficiency. An effect of aging has been observed in both dogs and cats: young dogs and cats (up to 2 years of age) have serum ionized calcium concentrations slightly higher than those reported in “adult” animals (Deniz and Mischke, 1995; Mischke et al, 1996).
ASSAY OF VITAMIN D CONCENTRATIONS
Measurement of vitamin D metabolites, although not commonly used in veterinary medicine, would occasionally be helpful in the diagnosis of calcium disorders. 25[OH] vitamin D (calcidiol) and 1,25[OH]2 vitamin D (calcitriol) are the metabolites of greatest clinical interest for detection of hypovitaminosis or hypervitaminosis D syndromes and in renal failure (Carothers et al, 1994). These metabolites are stable during refrigeration and freezing, but samples should not be exposed to light for any length of time. Both metabolites are chemically identical in all species, therefore receptor-binding assays or radioimmunoassays (RIAs) used for people are satisfactory for dogs and cats (Hollis et al, 1996). Young growing individuals have higher calcitriol concentrations than adults.
Calcitriol assays have demonstrated genetic errors of vitamin D metabolism and normal-to-increased concentrations in PHPTH, whereas patients with hypercalcemia of malignancy have levels that may be low, normal, or high. Dogs and cats with possible exposure to rat and mouse poisons containing vitamin D (e.g., Rat-B-Gone, Mouse-B-Gone, Rampage, Quintox) may present challenging diagnostic problems. Serum concentrations of vitamin D are increased in these toxicity states (Dougherty et al, 1990).
ASSAY OF PARATHYROID HORMONE–RELATED PROTEIN
Background
Parathyroid hormone–related protein (PTHrP) was identified in the 1980s as a tumor product that had the ability to activate PTH receptors and cause hypercalcemia. PTHrP resembles PTH not only in terms of its genetic sequence but also in terms of structure (Fig. 16-3), thus PTHrP is a second member of the PTH family of hormones (Broadus and Stewart, 1994). The first clues to the function of PTHrP came from studies of its expression in different tissues. In marked contrast to PTH, which is found only in the parathyroid glands, PTHrP is found in many tissues in both fetuses and adults, including epithelia, mesenchymal tissues, endocrine glands, and the central nervous system (Table 16-2). This widespread distribution suggests a diversity of roles for PTHrP (Fig. 16-4). Insights into these roles are now emerging from gene-knockout experiments and other approaches (Strewler, 2000). However, the key importance of this peptide is its central role in the development of humoral hypercalcemia of malignancy (HHM). It appears that the 1-36–amino acid peptide portion of PTHrP has an effect on cartilage, breast, and skin; the 38-94 midregion of PTHrP has an effect on placental calcium transport and perhaps on renal bicarbonate transport; the 107-139–amino acid portion of the molecule has an effect on the brain and on bone resorption (Strewler, 2000).
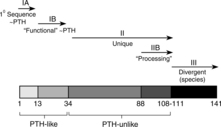
TABLE 16-2 SITES AND PROPOSED ACTIONS OF PARATHYROID HORMONE-RELATED PROTEIN (PTHrP)
| Site | Proposed Actions | |
|---|---|---|
| Mesenchymal Tissues | ||
| Cartilage | Promotes proliferation of chondrocytes; inhibits terminal differentiation and apoptosis of chondrocytes | |
| Bone | Stimulates or inhibits bone resorption | |
| Smooth muscle | Released in response to stretching; relaxes smooth muscle | |
| Vascular system | ||
| Myometrium | ||
| Urinary bladder | ||
| Cardiac muscle | Positive chronotropic stimulus; indirect positive inotropic stimulus | |
| Skeletal muscle | Unknown | |
| Epithelial Tissues | ||
| Mammary | Induces branching morphogenesis; secreted in milk; possible roles in lactation | |
| Epidermis | Unknown | |
| Hair follicle | Inhibits anagen | |
| Intestine | Unknown | |
| Tooth enamel | Induces osteoclastic resorption of overlying bone | |
| Endocrine Tissues | ||
| Parathyroid glands | Stimulates placental transport of calcium? | |
| Pancreatic islets | Stimulates insulin secretion and somatic growth | |
| Pituitary | Unknown | |
| Placenta | Calcium transport? | |
| Central nervous system | Released from cerebellar granular neurons in response to activation of L-type calcium channels; receptors in cerebellum, hippocampus, hypothalamus | |
From Strewler, 2000.

PTHrP Assay Systems
Two-site IRMA and N-terminal RIAs are available for the measurement of human PTHrP, and these same assay systems have been validated for the dog because of the high degree of sequence homology between species (Brown et al, 1987; Burtis, 1992). PTHrP is susceptible to degradation by serum proteases, therefore these assays should be run from fresh or frozen plasma using EDTA as anticoagulant. The EDTA complexes with plasma calcium, limiting the action of most proteases. The addition of protease inhibitors, such as aprotinin, may provide further protection. Use of serum is not recommended (Rosol et al, 2000).
Clinical Usefulness
Simultaneous measurement of PTH and PTHrP in the plasma may be especially valuable when attempting to distinguish primary hyperparathyroidism (PHPTH) from hypercalcemia of malignancy (HHM) (Burtis et al, 1990). In several studies, the plasma PTHrP concentration was increased in most people with HHM, including those with metastatic disease. PTHrP has been detectable in only a small percentage of humans with primary hyperparathyroidism, this hypercalcemia being attributed to increased PTH concentrations. Impaired renal function is associated with increased immunoreactivity to PTHrP fragments (Budayr et al, 1989; Henderson et al, 1990).
There seems to be little doubt that valid assays for dog and cat PTHrP serve as an excellent tumor marker. In other words, simultaneous assaying for PTH and PTHrP in hypercalcemic dogs and cats has the potential to be a quick, reliable, noninvasive, and inexpensive means of distinguishing between malignant and nonmalignant hypercalcemia. However, animals with renal insufficiency may also have increased concentrations of PTHrP without malignancy (see Fig. 16-2).
DIFFERENTIAL DIAGNOSIS OF HYPERCALCEMIA
TABLE 16-3 DIFFERENTIAL DIAGNOSES OF HYPERCALCEMIA
Hypercalcemia of Malignancy
GENERAL OVERVIEW.
Malignancy-associated hypercalcemia is the most common cause of increased serum calcium concentrations in dogs and cats. Neoplasms can cause hypercalcemia by three recognized physiologic processes (Fig. 16-5): (1) humoral hypercalcemia of malignancy (HHM); (2) hematologic malignancies growing in the bone marrow; and (3) hypercalcemia induced by metastases of solid tumors to bone (Rosol et al, 2000). Dogs with lymphosarcoma, apocrine gland carcinomas of the anal sac, and multiple myeloma frequently have hypercalcemia. Some of the tumors less commonly associated with hypercalcemia include malignant mammary tumors, nasal adenocarcinomas, thyroid carcinoma, thymoma, squamous cell carcinoma of the gastrointestinal system or vagina, and melanoma.
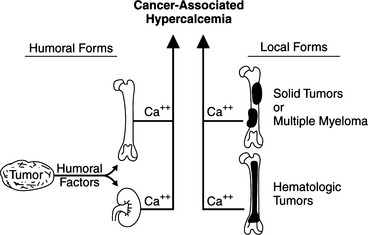
HUMORAL HYPERCALCEMIA OF MALIGNANCY
Humoral “Factors.”
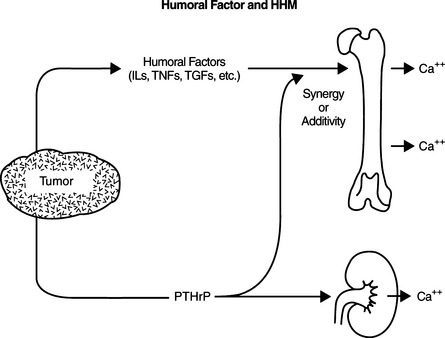
Tumor tissue at a site distant from bone may synthesize PTHrP, which is released into the circulation, stimulating bone resorption and causing hypercalcemia. Excessive secretion of biologically active PTHrP plays a central role in the pathogenesis of hypercalcemia in most forms of HHM. Cytokines, such as interleukin-1 (IL-1), tumor necrosis factor-α (TNF-α), transforming growth factor-α (TGF-α), transforming growth factor-β (TGF-β), or calcitriol, can have synergistic or cooperative actions with PTHrP (Fig. 16-6) (Rosol et al, 2000). PTHrP binds to the N-terminal PTH-PTHrP receptor in bone and kidney but does not cross-react immunologically with native PTH. The PTHrP stimulates osteoclastic bone resorption, increases renal tubular calcium resorption, and decreases renal tubular phosphate resorption. IL-1 and the transforming growth factors also have the potential to stimulate bone resorption (McCauley et al, 1991; Rosol et al, 2000). Not only does PTHrP provide the physiologic explanation for humoral hypercalcemia, it also explains why some tumors are not associated with hypercalcemia. Furthermore, assays for PTHrP are highly touted as tumor markers in that abnormal concentrations are obtained only from individuals with malignancies and renal failure (see Fig. 16-2).
Hematopoietic Neoplasia.
Lymphosarcoma is the most common hematopoietic tumor and the most common cause of hypercalcemia in dogs. It afflicts dogs of any age and either sex. Clinical signs may be subtle, moderate, or severe, or these dogs can be terminally ill. Approximately 20% to 40% of dogs with lymphosarcoma are hypercalcemic (Weller et al, 1982; Matus et al, 1986; Rosol et al, 2000). Hypercalcemia has been documented in cats with lymphosarcoma, but the incidence is much lower than in dogs. Studies on affected dogs have demonstrated parameters consistent with influence by PTHrP, including increased fractional excretion of phosphorus, increased nephrogenous cAMP, and increased osteoclastic bone resorption. PTHrP appears to differ from natural PTH in its inability to stimulate renal formation of 1,25 [OH]2 vitamin D and its lack of cross-reactivity with specific, two-site, intact PTH assays. A significant percentage of these dogs have increased serum concentrations of PTHrP (Fig. 16-7) (Weir et al, 1988a, 1988b, and 1988c).
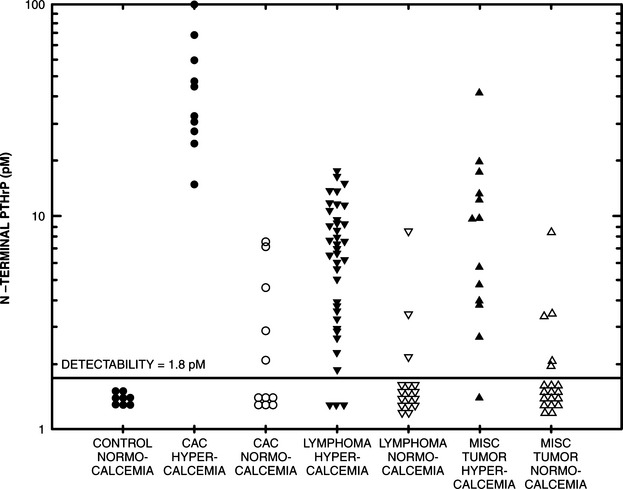
Lymphomas can synthesize both PTHrP and vitamin D, in which case the dog would not only have the expected increases in serum PTHrP but also would have increased vitamin D concentrations (Seymour and Gagel, 1993). Some lymphocytes contain the 1α-hydroxylase (similar to that found in renal tubules) that converts 25-hydroxyvitamin D to the active metabolite, 1,25-[OH]2 vitamin D (Rosol et al, 2000). Therefore lymphomas that retain this capability may synthesize excessive amounts of calcitriol, which would increase calcium absorption from the intestinal tract and exacerbate the hypercalcemia caused by PTHrP (see Fig. 16-2).
In contrast to the persistent hypercalcemia observed in dogs with PHPTH, dogs with malignancy-associated hypercalcemia may have fluctuating serum concentrations of calcium and sometimes are normocalcemic. Although the clinical signs associated with lymphoma are variable, it is safe to state that dogs with a combination of lymphoma and HHM, as a group, are considerably more ill than dogs with PHPTH. However, until proven otherwise, lymphoma must remain a possible explanation for hypercalcemia in any dog or cat. Lymphosarcoma may or may not be apparent to the veterinarian during the physical examination. At least 40% of dogs with both lymphoma and HHM do not have peripheral lymph node, liver, spleen, or renal enlargement. Most of these dogs have an anterior mediastinal mass that is usually obvious on thoracic radiographs. The finding of an anterior mediastinal mass in a dog with hypercalcemia should suggest a diagnosis of lymphoma, although other forms of neoplasia (e.g., thymoma) may account for both the mass and the hypercalcemia (Foley et al, 2000).
Apocrine Gland Adenocarcinoma of the Anal Sac.
Adenocarcinomas of the anal sac represent another classic example of a cancer commonly associated with hypercalcemia. Like lymphosarcoma, this neoplasm is known to synthesize PTHrP (Matus and Weir, 1989). This is a well-defined but relatively uncommon tumor, and the diagnosis is usually made in older dogs; the mean age at presentation is 10 to 11 years, but the range is 3 to 17 years (Bennett et al, 2002). Although early reports describe this neoplasia primarily in females, no obvious gender predilection was noted in recent studies. In a review of 238 dogs with this condition, numerous breeds were represented; in another report, mixed breed dogs were most commonly afflicted (Ross et al, 1991; Goldschmidt and Shofer, 1992; Bennett et al, 2002). Clinical signs associated with this condition include (among many owner observations): recognition of a mass near the rectum, tenesmus, poor appetite or anorexia, polyuria/polydipsia, and lethargy.
In dogs with polyuria/polydipsia, hypercalcemia was usually present. However, not all hypercalcemic dogs had evidence of polyuria/polydipsia (Bennett et al, 2002). The incidence of hypercalcemia has varied in different studies from about 25% of afflicted dogs to about 50% (Ross et al, 1991; Bennett et al, 2002). In one study, the serum calcium concentrations (upper reference value of 12.6 mg/dl) at the time of diagnosis ranged from 12.7 to 21.7 mg/dl (Bennett et al, 2002). Tumor resection results in normocalcemia. Local reappearance of the cancer or metastasis causes recurrence of hypercalcemia. Dogs afflicted with this form of cancer have increases in urinary cAMP and fractional phosphorus excretion. Serum PTH concentrations have been suppressed, and bone histomorphometry has revealed increased bone resorption with no compensatory increase in formation. These observations are consistent with the biologic actions of PTHrP (Meuten et al, 1983b). Apocrine gland adenocarcinoma cell lines established in mice have also demonstrated that of the potential factors responsible for HHM, only PTHrP is increased in the serum (Grone et al, 1998). Successful treatment of this neoplasia results in rapid normalization of serum calcium, phosphorus, calcitriol, and PTH concentrations (Rosol et al, 1992b).
Other Nonneoplastic and Solid Tumors that May Synthesize PTHrP.
Interstitial cell tumors of the testicle, squamous cell carcinoma, thymoma, melanoma, and thyroid adenocarcinoma are less commonly encountered solid tumors that cause hypercalcemia without bone metastasis (Grain and Walder, 1982; Meuten et al, 1983a; Pressler et al, 2002). This underscores the value of PTHrP assays in dogs with hypercalcemia. The remission of hypercalcemia after tumor excision and the return of hypercalcemia after tumor recurrence or growth of metastases support the presence of a hypercalcemic factor (PTHrP) secreted by these tumors, but studies are limited. In humans, other tumors that may produce PTHrP include bronchogenic non–small-cell carcinomas, breast cancer, squamous cell carcinoma of the esophagus, renal-cell carcinoma, and hepatoma (Harris et al, 2002). PTHrP has also been associated with hypercalcemia in nonneoplastic diseases, such as schistosomiasis (Fradkin et al, 2001).
HEMATOLOGIC MALIGNANCIES GROWING IN THE BONE MARROW: OSTEOLYTIC HYPERCALCEMIA
Background.
Some types of hematologic malignancies in the bone marrow produce hypercalcemia by inducing bone resorption locally (Rosol et al, 2000). This condition is usually associated with lymphoma and multiple myeloma. In humans, metastatic breast cancer is another example, although this association is not common in dogs or cats. A number of paracrine factors, or cytokines, may be responsible for the stimulation of bone resorption in dogs with such tumors. The cytokines most often implicated in the pathogenesis of local bone resorption are IL-1 and IL-6, TNF-α and TNF-β, TGF-α and TGF-β, and PTHrP (Black and Mundy, 1994). Production of small amounts of PTHrP by a tumor in bone may stimulate local bone resorption without inducing a systemic response. Prostaglandins (especially prostaglandin E2) may also contribute to local stimulation of bone resorption (Rosol et al, 2000). Together, these cytokines and prostaglandins comprise the osteoclast-activating factors.
Multiple Myeloma.
Multiple myeloma is a tumor of the B-lymphocyte or plasma cell line that may be associated with the development of osteolytic bone lesions and, occasionally, hypercalcemia. The hypercalcemia develops secondary to production of osteoclast-activating factors. In addition, there is a correlation between the extent of bone destruction, the amount of tumor cell burden, and the amount of osteoclast-activating factor produced by myeloma cell cultures (Durie et al, 1981). Approximately 17% of dogs afflicted with this cancer are hypercalcemic (Matus et al, 1986), and 50% of dogs with multiple myeloma have radiographic evidence of bone lysis.
Bone pain is usually associated with the lytic areas. The initial database from these dogs often reveals abnormal increases in the total serum protein concentration as a result of a monoclonal spike in the globulin fraction. This monoclonal gammopathy can be demonstrated specifically with serum protein electrophoresis. Bone marrow aspiration confirms the diagnosis in most cases, demonstrating an infiltration of malignant plasma cells. Analysis of urine for light chains of myeloma protein (Bence Jones protein) has not been of much value.
HYPERCALCEMIA INDUCED BY METASTASES OF SOLID TUMORS TO BONE.
Certain malignant neoplasms with osseous metastasis may cause hypercalcemia and hypercalciuria. Primary bone tumors, by contrast, do not commonly induce hypercalcemia. For example, hypercalcemia would be rare in a dog with osteosarcoma. In contrast, malignant mammary adenocarcinoma or squamous cell carcinoma is infrequently associated with both bone metastasis and hypercalcemia. Several different types of cells may be involved in the actual destruction of bone at sites of metastasis, including osteoclasts, tumor cells, lymphocytes, and monocytes. Lymphocytes and monocytes may accumulate as part of the cell-mediated immune response to a tumor (Mundy et al, 1984). Osteolysis is a result of the physical disruption of bone by proliferating neoplastic cells, but it also can be caused by secretion of cytokines or prostaglandins that stimulate local bone resorption (Garrett, 1993).
Epithelial tumors, especially squamous cell carcinomas, are the most likely neoplasms to metastasize to bone in dogs and cats (Quigley and Leedale, 1983). Common metastatic bone sites in the dog include the humerus, femur, and vertebrae, whereas in the cat, local invasion of bone rather than distant metastasis is more common. Although reported, these tumors are not commonly associated with hypercalcemia (Grain and Walder, 1982).
Hypervitaminosis D and Rodenticide Toxicosis
BACKGROUND.
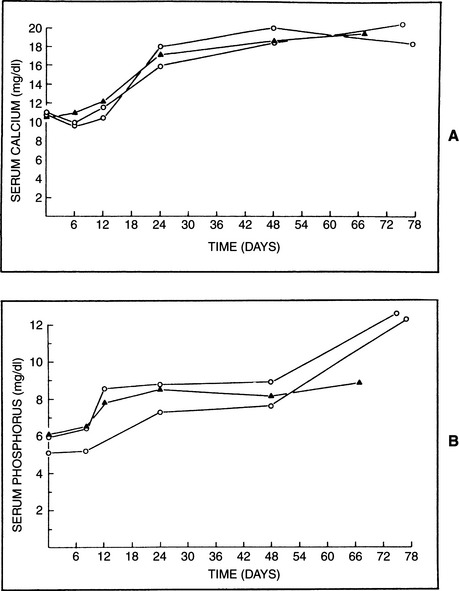
Vitamin D is cumulative in its action and may require 1 to 2 weeks before its maximum effects on mineral metabolism occur. However, acute toxicity after massive ingestion of cholecalciferol appears to be the more common experience when dealing with this problem in dogs. Hypercalcemia and hyperphosphatemia are the anticipated electrolyte abnormalities in animals with vitamin D toxicity, although normophosphatemia and transient periods of normocalcemia have been reported (Figs. 16-8 and 16-9) (Harrington and Page, 1983). Hypercalcemia begins as soon as 12 to 18 hours after ingestion, and peak concentrations are usually demonstrated by 48 to 72 hours, coinciding with increases in the BUN and creatinine (Rumbeiha, 2000). Increased resorption of bone, coupled with increased gastrointestinal absorption of calcium and phosphorus, is responsible for these abnormalities. Skeletal disease is usually not detectable radiographically, probably because of the acute nature of this form of toxicosis. The osteoclastic phase of bone resorption occurs early and is followed by osteoid deposition and hyperosteoidosis (Boyce and Weisbrode, 1983). Extensive soft tissue mineralization of the endocardium, blood vessels, tendons, kidney, and lung is frequently associated with vitamin D toxicity (Meuten, 1984).
RODENTICIDE TOXICOSIS.
Hypercalcemia that develops secondary to cholecalciferol rodenticide toxicosis in dogs and cats is a recognized concern (Gunther et al, 1988; Moore et al, 1988; Fooshee and Forrester, 1990; Dougherty et al, 1990). Rat bait products containing cholecalciferol include Quintox, Rampage, and Muritan, among other proprietary names (Rumbeiha, 2000; Morrow, 2001). Dogs studied after being given this type of poison became weak, lethargic, and anorexic within 48 hours. Within 60 to 70 hours of consumption, all dogs became recumbent, exhibited hematemesis, and progressed into shock before dying or being euthanized (Gunther et al, 1988). Although the median lethal dose of cholecalciferol in dogs is widely reported to be 43 to 88 mg/kg, studies have shown that as little as 10 mg/kg given once orally can be lethal. Dogs that ingest as little as 4 to 6 mg/kg once become ill. Clinically healthy dogs that ingest single doses of 2 mg/kg develop hypercalcemia (Rumbeiha, 2000).
Most dogs and cats exposed to these toxins have had rapid increases in the serum calcium and phosphate concentrations (see Fig. 16-8). Diffuse hemorrhage in the stomach and small intestine was obvious. Histologic lesions consisting of hemorrhage or mineralization or both were identified in the gastrointestinal tract, kidneys, and myocardium and in the blood vessels of many organs (Gunther et al, 1988). The incidence of acute and/or severe renal failure was variable. Three exposed cats survived (Moore et al, 1988).
Hypoadrenocorticism
Hypoadrenocorticism (Addison’s disease) is one of the more common causes of hypercalcemia in dogs, accounting for approximately 10% to 50% of cases (Uehlinger et al, 1998; Rosol et al, 2000). Serum calcium concentrations have been reported to be increased in as many as 33% of dogs with adrenocortical insufficiency (hypoadrenocorticism) (Peterson and Feinman, 1982; Peterson et al, 1996) as well as in a smaller percentage hypoadrenal cats (Johnessee et al, 1982; Peterson et al, 1989). In our series of hypoadrenal dogs, a correlation has been noted between the degree of hyperkalemia and the level of hypercalcemia (see Chapter 8). If the serum potassium concentration exceeds 6.0 to 6.5 mEq/L, a large percentage of these animals have serum calcium concentrations of 12 to 13.5 mg/dl. The hypercalcemia is not restricted to the extremely ill hypoadrenal dog. It is not common, however, for the serum calcium concentration to exceed 13.5 mg/dl, and it rarely exceeds 15 to 16 mg/dl. Despite the increased total serum calcium concentrations, serum ionized calcium levels usually remain in the reference range. Serum phosphate concentrations also correlate with serum calcium concentrations, with the hyperphosphatemic animal more likely to exhibit hypercalcemia (see Fig. 16-9).
Clinical signs and laboratory abnormalities associated with hypoaldosteronism (a primary component of Addison’s disease) usually are striking and overshadow concerns related to hypercalcemia (see Chapter 8). Most dogs and cats with hypoadrenocorticism have hyperkalemia, hyponatremia, azotemia, hyperphosphatemia, and related problems. The only differential diagnoses for this combination of serum abnormalities are hypoadrenocorticism, renal failure, and vitamin D (rodenticide) toxicosis. Furthermore, the hypercalcemia rapidly resolves after treatment for the adrenal insufficiency, frequently within a few hours of initiation of saline resuscitation.
The pathogenesis of the hypercalcemia associated with hypoadrenocorticism is probably multifactorial. Any combination of the following may be involved: (1) hyperproteinemia resulting from dehydration and hemoconcentration; (2) increased plasma protein– binding affinity for calcium; (3) increased concentrations of calcium-citrate complexes; and (4) increased renal tubular resorption of calcium (Peterson and Feinman, 1982).
Chronic Renal Failure
PATHOGENESIS OF HYPERCALCEMIA.
The pathogenesis of hypercalcemia associated with chronic renal failure is complicated. Diffuse hyperplasia of the parathyroid glands is characteristic of chronic renal failure. Parathyroidectomy decreases the serum calcium and PTH concentrations in dogs with hypercalcemia and renal failure (Meuten and Armstrong, 1989). The development of hypercalcemia in experimentally nephrectomized dogs depends on the presence of the thyroparathyroid complex (Tuma and Mallette, 1983). The kidneys excrete PTH and its metabolic byproducts, a process impeded by renal failure. Additional possible mechanisms for hypercalcemia in renal failure include (1) decreased renal excretion of calcium due to a reduction in the GFR; (2) increased concentrations of PTH due to both excessive secretion and reduced renal tubular hormone degradation, which could enhance bone resorption; (3) renal failure or PTH-induced increased concentrations of organic cations (e.g., citrates), which would increase complexed calcium concentrations; (4) an exaggerated response to vitamin D, with increased intestinal absorption of calcium; and (5) autonomous parathyroid gland activity (tertiary hyperparathyroidism), causing enhanced secretion rates (Meuten and Armstrong, 1989). The actual presence or incidence of a syndrome involving “autonomously functioning” parathyroid glands that are the result of chronic stimulation due to the renal disease (i.e., tertiary hyperparathyroidism) is not known. Tertiary hyperparathyroidism is the name given to the syndrome of chronic renal secondary hyperparathyroidism, in which one or more of the parathyroid glands begin to autonomously secrete PTH (“tertiary” disease).
Transient hypercalcemia may develop after administration of oral phosphate-binding agents, presumably as a result of shifts in calcium from bone to extracellular fluid in response to the rapid lowering of the circulating phosphorus concentration (Capen and Martin, 1983).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


