Mary Duncan
Gout in Exotic Animals
Historically, gout has been thought of as inflammatory arthritis that has developed in response to the deposition of urate crystals in the tissue (crystalline arthropathy). Differences have developed in the way the disease is reported in the veterinary and human medical literature, which bears further consideration in the future treatment and care of cases of gout in exotic species. In reptiles, generally, the same small cluster of cases forms the basis of suggested treatments in standard texts. From the position of a pathologist, the condition seems more common than these texts suggest—that cases frequently develop insidiously and that medical treatment is rarely successful.
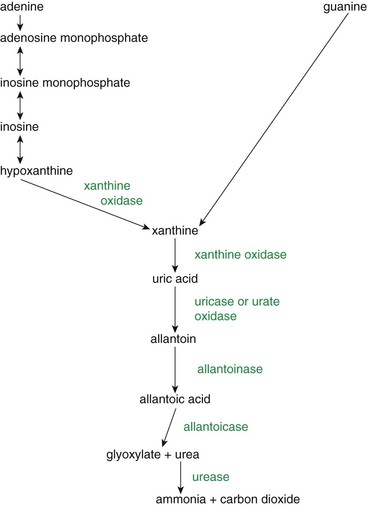
Urate is derived from the breakdown of purine bases (guanine and adenine), which may originate from nucleic acids of both exogenous and endogenous sources. Dietary nucleic acids (exogenous source) are broken down by pancreatic nucleases into nucleotides, which, in turn, are hydrolyzed to release pyrimidine and purine bases. Mucosal enzymes remove the phosphates and sugars.4 The degree of purine breakdown varies with species (Figure 67-1 and Box 67-1). Hypoxanthine produced by adenine breakdown may be excreted or converted to xanthine by xanthine oxidase, whereas all guanine breakdown results in xanthine generation, leading to uric acid production (see Figure 67-1). In humans, nonhuman primates, birds, some terrestrial reptiles, amphibians, and insects, uric acid is formed on purine breakdown. Urate salts and uric acid are relatively insoluble. With the enzyme uricase or urate oxidase, other mammals and reptiles break down urates further to allantoin, which is soluble.13 Guanine is excreted directly by pigs and spiders. Aquatic turtles excrete urea or ammonia. In fish, allantoin is further degraded into allantoic acid and urea. Aquatic invertebrates break down purines to ammonia. Excretion of both ammonia and urea result in the concomitant loss of water, so it is the form of excretion generally seen in semi-aquatic and aquatic species.
Amino acids are also assembled into purine and pyrimidine in the liver for the synthesis of nucleic acids (endogenous nucleic acid production). (Endogenous nucleic acid production is reduced in starvation.) When adenosine triphosphate (ATP) is used to generate energy, adenosine compounds are released.4 The adenosine is degraded via adenine to uric acid. Net degradation of ATP occurs in acute severe disease events such as infarction or severe tissue hypoxia, seizures, and tumor lysis syndrome (and, in humans, adult respiratory distress syndrome).4 Fructose phosphorylation in the liver requires abundant ATP; thus, diets high in fructose increase the risk of gout development and cause phosphate depletion.4 Fructose is the only carbohydrate that has a direct effect on urate metabolism.4,5 Oxidative phosphorylation is impaired in insulin-resistant individuals, so systemic adenosine levels rise with increased levels of intracellular coenzyme A esters and retention of sodium, urate, and water in the kidney.4
Two thirds of urate excretion from the body occurs through the kidneys and the remaining third from the gut.4 Circulating uric acid (monosodium urate) is filtered out of blood in the renal glomerulus, but nearly all the urate is resorbed in the proximal convoluted tubule, only to be actively secreted back into urine. The importance of the proximal convoluted tubule as a site of drug action becomes clear through these processes. Postsecretory reabsorption of urate is sodium dependent and stimulated by antiuricosuric drugs. (Insulin increases urate reabsorption by stimulating a urate anion exchanger; i.e., it drives circulating uric acid levels up, and the glycosuria seen in diabetes is uricosuric and may reduce the risk of gout.)13 Urate excretion is altered with renal disease, hypertension, and starvation. (Estrogen has an uricosuric effect and results in higher rates of gout in men than premenopausal women.)13
Damage to the renal tissue generally results in increased urate retention, and decreased perfusion leads to decreased urate clearance, for example, with dehydration or renal disease.9 Nephrotoxic drugs (aminoglycosides, sulfonamides) decrease urate excretion by damaging the renal tissue. The relative decrease in the glomerular filtration rate (GFR) that occurs in hypertension results in decreased urate excretion and increased risk of gout.4 Diuretics, alcohol intake, and acute diarrhea reduce hydration and so decrease urate excretion.13 Liver failure decreases the effectiveness of the filtering of blood and results in increased circulating urate levels. In humans, cases of gout are seen in association with diabetes, insulin resistance syndrome, cardiovascular disease, hypertension, nephropathy, and diseases with increased cell turnover (i.e., neoplasia and tissue breakdown), including tumor lysis disease, which is seen in primary leukemias, and lymphoma, and other malignancies.4,5 An increase in the incidence of gout in humans has been suspected in recent decades and is thought to have coincided with the rise in obesity and insulin resistance syndrome, dietary changes, and an aging population.4,13
Etiology
Primary gout occurs with overproduction of uric acid as an innate metabolic problem, which is generally caused by a defect in the renal urate transporters. Primary gout is the most common form seen in humans and usually has a familial inheritance (e.g., superactivity of the enzyme 5′-phosphoribosyl 1-pyrophosphate synthetase, deficiencies of hypoxanthine-guanine phosphoribosyl transferase, adenine phosphoribosyltransferase or xanthine dehydrogenase).1,4 An elevated predisposition to gout in the presence of other risk factors such as increased dietary purine is noted in certain groups (e.g., the Maori of New Zealand).1 Genetic factors resulting in hyperuricemia have not been identified in most urate-excreting species, although certain strains of poultry have been identified to have a simple autosomal recessive gene that causes a defect in the renal tubular secretion of urate.14
During evolution, gene mutations in primates resulted in variable activity of the uricase enzyme, depending on species.1 The retention of these gene mutations suggests that the loss of uricase confers some advantage.1 Urate may act as a bloodborne antioxidant, removing singlet oxygen and free radicals.1,4,5 Protection against oxidative damage may be especially important in the nervous system;5 the metabolic rate of the brain is relatively high, and the higher levels of fatty acids and lipid in the tissue suggest a greater need for defense against lipid peroxidation.1 Hyperuricemia appears to be protective against some neurodegenerative diseases: Alzheimer disease, Parkinson disease, and amyotrophic lateral sclerosis.1 Uric acid may be important in the maintenance of blood pressure when diets are very low in salt, acutely through stimulation of the renin–angiotensin system and chronically by producing sensitivity to salt by microvascular and interstitial renal disease.1 Conversely, in cases of persistent hyperuricemia, hypertension develops.1
Secondary gout develops with the loss of balance between uric acid production and excretion, resulting in net underexcretion and eventually hyperuricemia. Excess purine intake results when the expected proportion of purine in the diet fed exceeds the amount in the natural diet for a particular species; the most common situation that arises is that of herbivorous reptiles fed a meat-rich diet. In general meat, fish, or shellfish diets have higher levels of purine compared with vegetable-based diets.3 Availability of water and hydration of the individual further affect purine levels (use of diuretics increases propensity to develop hyperuricemia).13 The diet provided may have a different percentage of dry matter from that in food naturally available to the species. The bioavailability of purines may vary in the diet of a captive animal relative to the form of its presentation in nature. Exhibit humidity and temperatures may affect uric acid levels.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


