Chapter 10 GLUCOCORTICOID THERAPY
The products secreted by the adrenal cortex, or outer portion of the adrenal gland, are hormones necessary for normal metabolic function. Although it is possible for life to continue for a transient period of time in the complete absence of adrenocortical function, serious metabolic derangements usually ensue, and the capacity of the individual to respond to physiologic or environmental stress is completely lost. Chances of survival are critically compromised in any animal with severe adrenocortical hormone deficiency. Life may also be sustained despite excesses in adrenocortical secretion. Again, severe metabolic derangements ensue that interfere with chances for normal survival. The vital role of the adrenal cortex is due to the production and regulated secretion of a group of hormones, all steroid in nature. Two major groups of hormones are synthesized and secreted by the adrenal cortex: mineralocorticoids and glucocorticoids. Glucocorticoids are perhaps the most widely used therapeutic agents in veterinary practice.
THE CHEMISTRY OF GLUCOCORTICOIDS
More than 50 natural steroids are synthesized and secreted by the adrenal cortex, but only a few have identified significant biologic actions. All adrenocortical hormones are derivatives of the cyclopentanoperhydrophenanthrene nucleus (Fig. 10-1). These steroids include androgens, glucocorticoids, and mineralocorticoids. All of the adrenocortical steroids, except the androgens, contain 21 carbon atoms, an α,β-unsaturated ketone in ring A, and an α-ketol chain (-COCH2OH) attached to ring D. They differ in the extent of oxygenation or hydroxylation at carbon 11, 17, or 19. Depending on whether the predominant biologic effect is related to electrolyte and water metabolism or to carbohydrate and protein metabolism, the adrenocortical steroids are classified as mineralocorticoid or glucocorticoid, respectively.
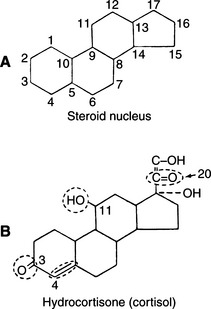
FIGURE 10-1 A, Steroid nucleus. B, Structure of cortisol. Circled structures are essential for glucocorticoid activity.
Clinical experience suggests that the antiinflammatory activity of adrenocortical steroids in humans correlates well with their glucocorticoid activity. The undesirable side effects (sodium retention, edema) are associated with mineralocorticoid activity. Synthetic steroids possessing higher glucocorticoid and lower mineralocorticoid activity than cortisol are commonly marketed. All adrenal corticoids require the 3-keto group and 4-5 unsaturation (see Fig. 10-1).
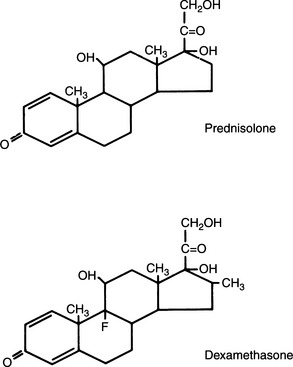
Additional unsaturation in ring A enhances antiinflammatory properties while reducing the sodium-retaining effect. Prednisolone, therefore, has four times the antiinflammatory activity of cortisol, yet only 0.8 the mineralocorticoid activity (Fig. 10-2). The presence of oxygen at position 11 is necessary for significant glucocorticoid activity but not for mineralocorticoid activity; the 11β-hydroxy group is more potent than the 11-keto group, which is converted into the active β-hydroxy group in the body. The 17α-hydroxy group is also important to glucocorticoid activity. The 21-hydroxy group is essential for mineralocorticoid activity; it favors but is not required for glucocorticoid activity. Introduction of either methyl or hydroxyl groups at position 16 markedly reduces mineralocorticoid activity but only slightly decreases glucocorticoid and antiinflammatory activity. Thus paramethasone (16α-methyl), betamethasone (16β-methyl), dexamethasone (16α-methyl), and triamcinolone (16α-hydroxy) have no significant mineralocorticoid activity (see Fig. 10-2). 6α-Methylation has unpredictable effects. It enhances the mineralocorticoid activity of cortisol but virtually abolishes that of prednisolone. The 9α-fluoro group enhances both glucocorticoid and mineralocorticoid activity, but the effect of substitutions at the 6 and 16 positions overrides this effect (Osol, 1980).
BIOLOGIC EFFECTS
Molecular Mechanisms
Once a steroid hormone has permeated a cell membrane, it binds with a cytosolic glucocorticoid receptor protein (Fig. 10-3). These proteins probably originate in the nucleus but migrate into the cytosol in the presence of steroids. After binding, the steroid-protein complex enters the cell nucleus and interacts with nuclear chromatin acceptor sites. The 90 kDa heat shock protein hsp90 may be involved in hormone-induced glucocorticoid receptor activity. The DNA binding domain of the receptor is a cysteine-rich region that assumes a conformation known as a “zinc finger” after chelating zinc. The receptor-glucocorticoid complex binds to specific sites in nuclear DNA, the glucocorticoid regulatory elements. This results in the expression of specific genes and the transcription of specific messenger ribonucleic acids (mRNAs). The resulting proteins elicit the glucocorticoid response, which may be inhibitory or stimulatory depending on the specific gene and tissue affected.
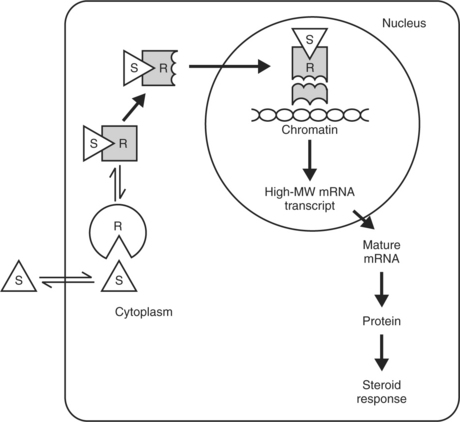
Although glucocorticoid receptors are similar in many tissues, the proteins synthesized in response to glucocorticoids vary widely and are the result of specific gene expression. The mechanisms under-lying this specific regulation are not known. Analyses of cloned complementary deoxyribonucleic acids (DNAs) for human glucocorticoid receptors have revealed marked structural and amino acid sequence homology between glucocorticoid receptors and receptors for other steroid hormones (e.g., mineralocorticoids, estrogen, progesterone), as well as for thyroid hormone and the oncogene v-erb A (Aron et al, 2001). Although the steroid-binding domain of the glucocorticoid receptor confers specificity for glucocorticoid binding, glucocorticoids such as cortisol and corticosterone bind to the mineralocorticoid receptor with an affinity equal to that of aldosterone. Mineralocorticoid receptor specificity is maintained by the expression of 11β-hydroxysteroid dehydrogenase in classic mineralocorticoid-sensitive tissues. The expression of this glucocorticoid-inactivating enzyme in other tissues may serve to protect those tissues from excessive glucocorticoid action (Aron et al, 2001).
Mechanisms distinct from those described here are likely responsible for unique glucocorticoid actions. The most significant example is that of glucocorticoid-induced “fast feedback” inhibition of ACTH secretion. This effect occurs within minutes of glucocorticoid administration, which suggests that it is not due to the typical induction of RNA and protein synthesis. Rather, the feedback is probably related to changes in secretory function or cell membranes (Aron et al, 2001).
Intermediary Metabolism
Glucocorticoids, in general, inhibit DNA synthesis. In addition, in most tissues they inhibit RNA and protein synthesis and accelerate protein catabolism. These actions provide substrate for intermediary metabolism; however, accelerated catabolism also accounts for the deleterious effects of glucocorticoids on muscle, bone, connective tissue, and lymphatic tissues. In contrast, RNA and protein synthesis in the liver are stimulated (Aron et al, 2001).
The effect of glucocorticoids on intermediary metabolism can be summarized as follows: (1) effects are minimal in the fed state; however, during fasting, glucocorticoids contribute to the maintenance of plasma glucose concentrations by increasing gluconeogenesis, glycogen deposition, and the peripheral release of substrate; (2) hepatic glucose production is enhanced, as is hepatic RNA and protein synthesis; (3) the effects on muscle are catabolic (i.e., decreased glucose uptake and metabolism, decreased protein synthesis, and increased release of amino acids); (4) in adipose tissue, lipolysis is stimulated; and (5) in glucocorticoid deficiency, hypoglycemia may result, whereas in states of glucocorticoid excess, there may be hyperglycemia, hyperinsulinemia, muscle wasting, and weight gain with abnormal fat distribution (Tyrrell et al, 1991).
Effects on Other Tissues
BONE.
The physiologic role of glucocorticoids in bone metabolism and calcium homeostasis is not well understood. However, in excess, they directly inhibit bone formation by decreasing cell proliferation and the synthesis of RNA, protein, collagen, and hyaluronate. Glucocorticoids also directly stimulate bone-resorbing cells, leading to osteolysis and increased urinary hydroxyproline excretion. They also potentiate the actions of parathyroid hormone (PTH) and 1,25-dihydroxycholecalciferol (1,25[OH]2D3) on bone, and this may further contribute to net bone resorption.
CALCIUM METABOLISM.
Glucocorticoids markedly reduce intestinal calcium absorption, which tends to lower the serum calcium concentration. This effect results in secondary increases in PTH secretion, in order to maintain the serum calcium concentration in the normal range by stimulating bone resorption. In addition, glucocorticoids may directly stimulate PTH release. The mechanism of decreased intestinal calcium absorption is unknown, although studies have demonstrated that it is not decreased synthesis or decreased serum concentrations of the active vitamin D metabolites; in fact, 1,25-vitamin D concentrations are normal or even increased in the presence of glucocorticoid excess. Increased 1,25-vitamin D synthesis may result from decreased serum phosphorus concentrations, increased serum PTH concentrations, and direct stimulation of renal 1α-hydroxylase. Glucocorticoids also increase urinary calcium excretion, and hypercalciuria is a consistent feature of cortisol excess in humans. Reduced tubular resorption of phosphate, leading to phosphaturia and decreased serum phosphorus concentrations, also occurs (Aron et al, 2001).
CENTRAL NERVOUS SYSTEM (CNS).
Glucocorticoids readily enter the brain, and although their physiologic role in CNS function is unknown, an excess or deficiency may profoundly alter behavior and cognitive function in humans (Tyrrell et al, 1991).
LEUKOCYTES.
Glucocorticoids influence both white cell movement and function. Under the influence of glucocorticoids, white blood cell counts increase due to an increased rate of neutrophil (polymorphonuclear neutrophil [PMN]) release from the bone marrow, an increased circulating half-life of PMNs, and by inhibiting PMN movement out of the vascular space. Glucocorticoids reduce the numbers of circulating lymphocytes, monocytes, and eosinophils by increasing their movement out of the circulation. Glucocorticoids also reduce migration of inflammatory cells (PMNs, monocytes, and lymphocytes) to sites of injury. This is probably the primary mechanism of the antiinflammatory action and increased susceptibility to infection associated with chronic steroid administration (Aron et al, 2001).
Effects on the Function of Endocrine Organs
IATROGENIC ADRENAL SUPPRESSION.
The hypothalamic-pituitary-adrenal (HPA) axis of healthy dogs can be suppressed for an average of 32 hours after one intravenous injection of dexamethasone at a dosage of 0.1 mg/kg. Smaller doses result in a shorter mean duration of HPA suppression, which demonstrates that this suppressive effect is dose dependent (Kemppainen and Sartin, 1984). Cortisol typically suppresses the HPA axis for approximately 12 to 24 hours, whereas prednisolone is suppressive for 12 to 36 hours (Table 10-1). Adrenal suppression can follow treatment with almost any form of glucocorticoid. Ocular and topical skin formulations are examples of steroids that can be administered for several days or longer, can be absorbed systemically, and subsequently can suppress the HPA axis for weeks (Roberts et al 1984; Zenoble and Kemppainen, 1987).
TABLE 10-1 DURATION OF ACTION OF NATURAL AND SYNTHETIC GLUCOCORTICOIDS FOLLOWING ORAL OR IV ADMINISTRATION
| Drug | Duration of Action |
|---|---|
| Short-Acting | |
| Hydrocortisone | <12 hr |
| Cortisone | <12 hr |
| Intermediate-Acting | |
| Prednisone | 12–36 hr |
| Prednisolone | 12–36 hr |
| Methylprednisolone | 12–36 hr |
| Triamcinolone | 12–36 hr |
| Long-Acting | |
| Betamethasone | >48 hr |
| Dexamethasone | >48 hr |
| Flumethasone | >48 hr |
| Paramethasone | >48 hr |
Glucocorticoids given for days to weeks rarely have any prolonged significant clinical effects, but more chronic administration of any corticosteroid could cause adrenocortical atrophy (iatrogenic hypoadrenocorticism). However, the degree of individual variation in response to glucocorticoid administration among dogs and cats is so pronounced that each dog and cat must be evaluated independently (Brockus et al, 1999).
Recovery of adrenocortical function occurs relatively quickly in dogs and cats after discontinuation of chronic steroid administration. Adrenocorticotropic hormone (ACTH) response test results usually return to normal within weeks. We almost never “taper” dogs or cats off steroids when administration of these drugs has been demonstrated to be unnecessary. Rather, the steroids are simply stopped. If there is concern about clinical signs caused by iatrogenic hypoadrenocorticism, steroids can be given at doses slowly tapered over a period of weeks, but this approach is almost never used. (Further discussion on suppression of the HPA axis is provided under Adverse Reactions, page 477.)
THYROID FUNCTION.
Although basal thyroid-stimulating hormone (TSH) concentrations in humans are usually normal, TSH responsiveness to thyrotropin-releasing hormone (TRH) is frequently subnormal after chronic glucocorticoid therapy. Serum total thyroxine (T4) concentrations are usually low-normal because of a decrease in thyroxine-binding globulin, but free T4 concentrations are normal. Both the total and free T3 concentrations may be low, because glucocorticoid excess decreases conversion of T4 to T3 and increases conversion to reverse T3. Despite these alterations, manifestations of hypothyroidism are not apparent (Aron et al, 2001).
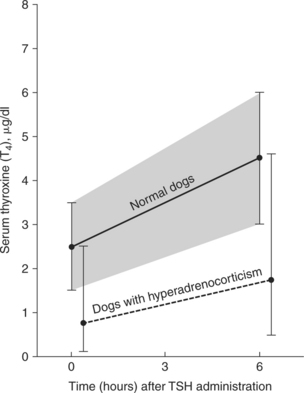
The serum total and free T4 concentrations may be low normal or decreased both in dogs treated with glucocorticoids and in those with naturally occurring hyperadrenocorticism. Serum T4 response to TSH may parallel but remain below normal reference limits (Fig. 10-4). The clinical and biochemical changes associated with chronic glucocorticoid excess in dogs are similar to those observed in hypothyroidism (lethargy, weakness, dermatologic alterations, hypercholesterolemia). However, none of these require direct therapy, because correcting the cortisol excess resolves these problems (Woltz et al, 1983).
Antiinflammatory Properties
OVERVIEW.
The capacity of glucocorticoids to prevent or suppress inflammatory reactions not only is grossly visible by limiting heat, redness, swelling, and tenderness, but also can be documented microscopically. Glucocorticoids inhibit the early inflammatory phenomena of edema, fibrin deposition, capillary dilation, migration of leukocytes, and phagocytic activity. Glucocorticoids also limit later manifestations of inflammation such as capillary proliferation, fibroblast proliferation, deposition of collagen and, still later, cicatrization (Gilman et al, 1980; Aron et al, 2001).
Glucocorticoids inhibit inflammation whether the inciting agent is radiant, mechanical, chemical, infectious, or immune mediated. Such therapy is palliative in that the underlying cause of a disorder may remain, but its clinical manifestations are suppressed. This ability to suppress inflammation, regardless of its cause, has made glucocorticoids valuable therapeutic agents. However, these encompassing properties also make glucocorticoid therapy dangerous, because it can mask the clinical expression of a disease process. The inflammation that allows the clinician to recognize and monitor any nonsteroidal course of therapy can be completely suppressed, allowing a disease to continue or worsen while preventing this process from being identified. As with any therapy, the benefits of glucocorticoid medication must be weighed against the risks of their use.
Relative to dogs and cats, the rabbit, rat, mouse, and human being are relatively more sensitive to the antiinflammatory and/or immunosuppressive effects of glucocorticoids. Thus the results of research completed in the last four species, on in vitro models, and even in healthy animals may not apply to dogs and cats with naturally occurring diseases (Papich and Davis, 1989).
With these caveats established, several general effects of glucocorticoids have been documented. Polymorphonuclear neutrophils demonstrate decreased migration and egress into inflammatory tissue. The circulation of T-cell lymphocytes is decreased, and lymphocyte activation is suppressed. Glucocorticoids decrease vascular permeability and also the rate of synthesis of prostaglandins, prostacyclin, thromboxane, and leukotriene (Papich and Davis, 1989).
NEUTROPHILS
Membrane Effects.
The classic antiinflammatory effect of glucocorticoids is decreased migration of PMNs into inflamed tissue. Soon after administration of a pharmacologic dose of glucocorticoids, neutrophilia occurs as a result of both increased release of cells from the maturation pool of the bone marrow and prolongation of PMN half-life (MacDonald, 2000). In addition, the egress of cells from blood vessels via diapedesis decreases. Because neutrophils mature within hours, inhibition of their egress results in a rapidly developing mature neutrophilia. The mechanism for decreased egress may be related to a cell surface configuration change that results in decreased vessel margination and adherence. Lysosomal membrane stabilization is probably a true antiinflammatory effect of glucocorticoids (Cupps and Fauci, 1982; Meuleman and Katz, 1985; Papich and Davis, 1989).
Phagocytosis.
Glucocorticoids may impair the ability of neutrophils to phagocytize and destroy bacteria. Steroids may also hinder microbicidal activity by decreasing oxidative metabolism, by decreasing fusion of neutrophil granules to lysosomes, or by affecting the pH in the lysosomes of phagocytes. Regardless of the mechanism, corticosteroids increase susceptibility to infection because of an impaired ability to kill bacteria (Papich and Davis, 1989).
LYMPHOCYTES.
The lymphocyte population is composed of a circulating pool and a noncirculating pool. The circulating pool freely moves between the intravascular compartment and the extravascular compartment (lymph nodes, spleen, bone marrow, and thoracic duct). After glucocorticoid administration, lymphocytes of the circulating pool redistribute to the extravascular compartment. T-cell lymphocytes are affected more than B cells (T cells constitute approximately 70% of the circulating pool). The discrepancy in the lymphocyte response is not easily explained, but the ability of B cells to metabolize glucocorticoids faster than T cells may be involved (Cupps and Fauci, 1982; Kehrl and Fauci, 1983; Papich and Davis, 1989). Furthermore, helper or inducer lymphocytes (CD4) are more sensitive than the cytotoxic suppressor cells (CD8). Steroids also inhibit the production of interleukin-2, resulting in inhibition of lymphocyte proliferation and cytotoxic function. Glucocorticoids also cause suppression of B-cell growth factors (BCGFs), affecting the proliferation of this cell type (MacDonald, 2000). Decreased numbers of circulating lymphocytes and decreased proliferative response of lymphocytes reduce these cells’ ability to participate in immunologic and/or inflammatory reactions. Additional observations include a decreased response to mitogens, suppressed lymphokine synthesis, and decreased transformation and antigen recognition (Cupps and Fauci, 1982; Meuleman and Katz, 1985; Papich and Davis, 1989).
Glucocorticoids have few direct effects on B cells, but they indirectly alter B-cell function by modulating accessory cells. A complex series of interactions between T cells, macrophages, antigens, and cell mediators is required for full expression of lymphocyte function (Nossal, 1987). Commonly used doses of glucocorticoids do not prevent an animal from mounting a normal immunologic response to vaccinations or other antigens (Meuleman and Katz, 1985), but as doses of corticosteroids increase, concentrations of immunoglobulins IgG, IgA and, to a lesser extent, IgM are decreased. Synthesis of IgE is not affected. Studies in humans suggest that repeated daily doses of long-acting corticosteroids, such as dexamethasone, suppress antibody synthesis but that alternate-day administration of antiinflammatory doses of an intermediate-acting steroid, such as prednisolone, does not (Papich and Davis, 1989). Glucocorticoids induce eosinopenia, which is most likely a consequence of changes in distribution to various tissues. Evidence for a lytic or toxic effect on eosinophils has not yet been proven (MacDonald, 2000).
MACROPHAGES.
Mononuclear cells have several critical functions. They serve as antigen-presenting accessory cells during the induction of cell-mediated and humoral immune responses. They also function as “professional” phagocytes against bacteria, fungi, and viruses. In combination with antibodies, they may have tumor cell–killing capacity. They are also essential in clearing senescent cells, particularly erythrocytes, and in bone remodeling, wound healing, and lung surfactant turnover (MacDonald, 2000).
Monocytes and macrophages (lymphocyte accessory cells) appear to be more sensitive to the effects of glucocorticoids than neutrophils. Therapeutic corticosteroid doses decrease phagocytosis and bactericidal activity. Steroids also diminish macrophage and monocyte ability to process antigens for presentation to B cells. Synthesis of cell mediators, such as interleukin-1 (IL-1), also may be suppressed. Corticosteroids inhibit both lymphokine synthesis and the cellular response to lymphokines (Cupps and Fauci, 1982; Dinarello and Mier, 1987; Papich and Davis, 1989). Glucocorticoids also inhibit production of tumor necrosis factor (TNF), thereby affecting a wide variety of inflammatory processes. The major antiinflammatory effect of glucocorticoids is related also to decreased accumulation of mononuclear phagocytes at inflammatory foci. This may be a consequence of decreased chemotactic factors by other cells, including T lymphocytes outside the mononuclear cell population. Antienzymatic effects of steroids may also be a factor. Inhibition of cytokines such as IL-1, interferon-γ (IFN-γ), and TNF/cachectin also has been documented (Cohn, 1991; Scott et al, 1995).
BASOPHILS AND MAST CELLS.
Glucocorticoids reduce the number of circulating basophils, most likely through reduced production. Glucocorticoids reduce the number of mucosal mast cells, although the subtype of mast cell found in connective tissue is less sensitive. The reduction of mast cells most likely is a result of inhibition of IL-3 or mast cell growth factor. A direct effect may also occur. Migration of basophils is significantly reduced by steroids. Some suggest that steroids directly inhibit production and release of mast cell inflammatory mediators, or that this may be the simple result of a smaller number of mast cells causing degranulation. Topical glucocorticoids decreasing response to antigen may also involve decreased cell numbers or decreased access of antigen to tissue mast cells (Butterfield and Gleich, 1989; Munck and Guyre, 1989).
ARACHIDONIC ACID METABOLISM.
In general, glucocorticoids induce an antiphospholipase effect by using second-messenger proteins called lipocortins. These proteins inhibit cellular phospholipase A, thus reducing the release of arachidonic acid and all subsequent proinflammatory mediators (prostaglandins, prostacyclines, thromboxanes, and leukotrienes). Lipocortins regulate cellular metabolism of phospholipases as well. Many cells release arachidonic acid when stimulated by neurotransmitters and certain drugs. Lipocortins are naturally digested by proteases. They promote the maturation of suppressor T cells and inhibit proliferation response to T cells by mitogens. β-Lipocortins have a dose-dependent inhibitory effect on the acute phase of cytotoxic T cells. They also inhibit antibody-dependent cytotoxicity by natural killer cells and protect cells from complement-mediated lysis (Cupps, 1989; Flower, 1989; Hirata, 1989).
EFFECTS ON BLOOD VESSELS.
The beneficial effects of glucocorticoids when administered to animals in shock, to animals with CNS edema, and to those with various other inflammatory conditions are related to “stabilization of microvascular integrity.” This phenomenon reduces edema formation and extravasation of cells from the vasculature to tissues. These effects may be attributed to suppressed neutrophil action and to reductions in prostaglandin synthesis. It has also been suggested that corticosteroids antagonize vasoactive substances such as histamine and kinins, thereby providing protection against the toxic effects of free oxygen radicals released during inflammation. Because it is known that macrophages release plasminogen activators as part of the cascade of reactions during inflammation, it is of interest that glucocorticoids likely inhibit this reaction. This is an important effect of glucocorticoids, because activated plasmin digests supporting tissue of vessels and enhances vascular permeability (Papich and Davis, 1989).
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


