CHAPTER 76 Genetic Testing in Domestic Cats
GENETIC STRUCTURE OF THE CAT
Understanding genetic testing in the feline species begins with understanding the domestic cat itself. The cat is a species that is domesticating itself, actively, right before our very eyes, with a little additional help of course from human beings. Cats represent one of the most recently domesticated animals, the domestication process likely starting within the past 5000 to 10,000 years, initiating around the time of agricultural development. Because the “wild” progenitor of the cat is relatively recent and because “domesticated” cats have been relatively “free-ranging” and uncontrolled, the genetic foundation of cat breeds is relatively diverse, perhaps more diverse than dogs but less diverse than human beings. Genetic diversity studies have shown that not all random-bred cats have the same genetics1,2 and that the origins of the cat may be important to the validity of a genetic test. Cats in Southeast Asia may have a completely different mutation for a disease than cats in the United States because their populations are quite genetically distinct.
A majority of the cats in the world are still represented by fairly free-ranging, random-bred individuals.3 Not only is cat domestication a recent event, but so too is the development of cat breeds.4–6 The cat breeds that were recognized at the initiation of the cat fancy in the late 1800s represented different geographical locales, and hence, different genetic populations of cats such as Siamese from Southeast Asia, Abyssinian from India, and Persians from Western Europe. These breeds remain genetically distinct today and represent the foundation of the cat fancy.1 However, most cat breeds have developed in the past 50 years, either as derivatives from these foundation breeds or from diverse random-bred populations. The different geographical origins of cat breeds imply highly distinctive genetics; thus, like the random-bred populations, genetic mutations found in cat breeds from Southeast Asia are not likely to be genetically in common (identical by descent) with genetic mutations of cats originating from the Mediterranean or Western Europe. Therefore the mutations that cause disease in one breed may not be the same mutation in all breeds, depending on when the mutation occurred in the cat domestication history or if cross-breedings have occurred.
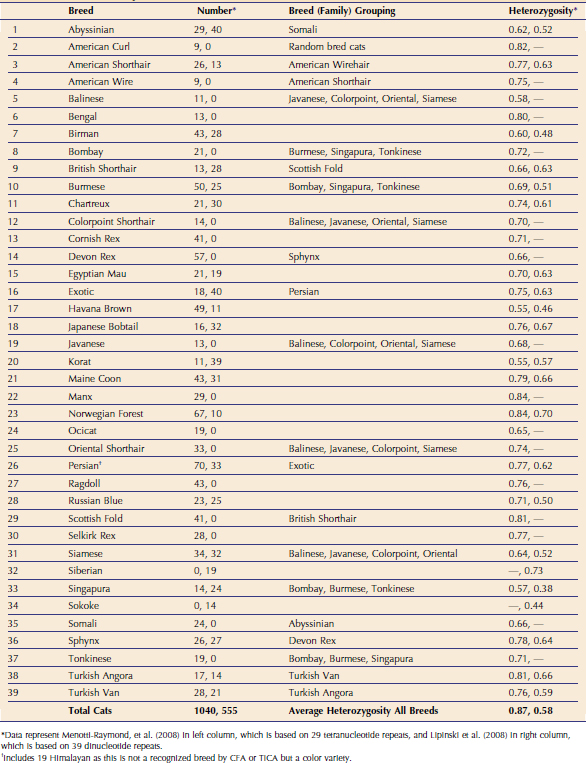
Two studies have analyzed the genetic makeup of cat breeds (Table 76-1).1,2 Menotti-Raymond et al evaluated 37 breeds, 21 of which were clearly distinct and 15 forming breed groups. Breeds such as Persian and Exotic could not be resolved as distinctly at the basic genetic level common as the other cat breeds. Analyses of these “breed families” managed to show sub-structuring, indicating that subbreeds can be defined in the cat. Lipinski et al found similar genetic demarcations of cat breeds, analyzing 22 breeds, identifying 19 distinct breed populations and three breed groups. In addition, the association of cat breeds with their geographical origins was identified. Thus knowing the association and relationship of cat breeds can help predict the likelihood that the same genetic mutation is causing a trait in one cat population versus another.
DOMESTIC CAT MUTATIONS
Some genetic mutations are quite old and inherent in the domestication process of the cat itself, such as many coat color mutations. The color mutations for solid coloration,7 brown variants,8 and dilution9 have shown to be the exact same mutation in all cat populations, east and west: likely an ancient single mutation event. However, the mutations for albinism probably occurred later in the domestication process because they have a higher prevalence in one geographical location, Southeast Asia.10 The albinism color variant that causes the Siamese and Burmese pointing pattern on the cat’s head, ears, paws, and tail likely originated in Southeast Asia and then spread around the world, once other cat breeds developed that desired the coloration.
Mutations that have occurred in one geographical group of cats can be spread to others by virtue of cross-breeding. Many newly developing breeds often incorporate older, well-established breeds to “short-cut” the time required to obtained desired colorations and morphological traits. One of the oldest breeds, the Persian, has been used in the production of American Shorthairs, Scottish Folds, and Selkirk Rex, quickly “borrowing” the genes responsible for brachycephalia, a desired head-type in each of these breeds. Along with the brachycephalia, these new breeds also suffer from polycystic kidney disease (PKD),11 a highly prevalent genetic disease in Persian cats.12–16 The use of Persians in several of these breeds is so intense and/or so recent that they can not be distinguished clearly as separate breeds on the overall genetic level. Even the Exotic Shorthair, a longstanding short-haired version of the Persian, can not be differentiated from a Persian based on genetic evaluations.1,2 Therefore the Exotic is just a single gene variety of a Persian. However, if cross-breeding is performed sparingly with established breeds, then newer developing breeds can become genetically distinct. The coloration of the foundation cat breed Siamese is found in many Western European and Mediterranean breeds, such as Ragdolls and Persians. However, the overall genetic structure of these newer breeds is distinctive from Siamese.
The genetics of long fur in the domestic cat is an interesting variation on a theme. At least four genetic mutations in the same gene, fibroblast growth factor 5 (FGF5), are causative for long fur in cats.17,18 Specific long fur mutations are more prevalent than others in particular breeds of cats, meaning some breeds have more than one mutation causing long fur. This implies that independent mutation events for long fur have occurred in the original population sources for a given breed. Therefore not all long-haired cats within a breed have the same mutation and some cats may be compound heterozygotes. A compound heterozygote is an individual who has two different mutations, but together those two mutations cause the individual to have the phenotype of interest. Long fur is a recessive trait, thus a cat with only one of any given mutation for long fur will still have short fur, but when two mutations are present, even though they are different mutations, the cat’s fur will be long. Therefore for a genetic test to identify carriers of long fur, all FGF5 mutations must be genotyped in the cats of interest. The same circumstance will occur when different mutations cause the same clinical disease, which is termed disease heterogeneity. Thus genetic testing laboratories must account for multiple mutations and this information must be conveyed to the veterinarian and owner appropriately.
Understanding cat breed relationships and disease heterogeneity helps to not only predict the causative factors of a disease, but also to clarify when genetic tests should be recommended and how they should be interpreted. As mentioned previously, the mutation causing PKD is found in most breeds that have used Persians as out-crosses. These breeds also show high similarity in their overall genetic composition. An alternative example is hypertrophic cardiomyopathy (HCM). A mutation for HCM has been identified in the Maine Coon breed19; however, Ragdolls have a different mutation in the same gene associated with the same type of cardiac disease20 (see Chapter 39). The two genetic forms of HCM are a perfect example of disease heterogeneity, a similar clinical presentation caused by different genetic factors. Although a few Ragdolls and Maine Coons may interbreed, overall, the crossing of the two breeds is not a common or acceptable practice. Therefore two breeds, both developed from random-bred cats of the United States and not as derivatives of one another, have two unrelated mutations for HCM, even though these mutations are in the same gene. A variety of other breeds and cat populations, such as Siberians and Bengals, are noted to have a prevalence of HCM.21–25 Only time will tell if their causative mutations are shared with Ragdolls and Maine Coons because of interbreeding, or if each breed has had the “fickle finger of fate” alter their genetic material in a unique manner. Overall, a genetic mutation must be proven to be causative in each breed by considering population histories and by confirming that the disease is completely correlated with the known genetic mutations.
TYPES OF GENETIC MUTATIONS
A variety of methods are used to implicate specific gene candidates for a particular trait of interest. However, proving the causative action of a mutation can be even more complicated. Comparative genetics, using the knowledge of causative genes and mutations from other species with similar traits and applying this knowledge to the species of interest, is a long-standing technique and often is supportive evidence for declaring a genetic mutation as causative for a trait. Deoxyribonucleic acid (DNA) mutations causing several inborn errors of metabolism, such as gangliosidosis26–30 or the various types of mucopolysaccharidosis,31–34 were readily identified by comparative genetic approaches. However, the normal or “wild-type” presentation for any trait or condition can be altered at the genetic level in a variety of manners.
One of the simplest alterations of a gene is termed a single nucleotide polymorphism (SNP). Some SNPs are innocuous and never affect the gene and its protein and are general polymorphisms or DNA variants. In the case of a disease-causing SNP, the changed nucleotide needs to alter the genetic code in a manner that will affect the protein produced by that gene. An SNP can cause an amino acid to change to a code that causes the termination of protein construction, a stop codon. The mutation of PKD changes the nucleotide sequence of the gene, which causes nearly a third of the normal protein not to be made during protein synthesis.11 The genomic sequence of a gene is divided into regions that code for the protein sequence, called exons, which are separated by noncoding sequences, called introns. The exons must be all spliced together to allow a normal protein to be synthesized. Some SNPs can alter the nucleotide signals that are at the beginning and ends of exons and introns, leading to exon skipping and exon splicing errors. A mutation affecting the exon-intron junction in tyrosinase-related protein (TYRP) causes alterations in the coat coloration of the cat.8 Mutations that cause stop codons and splicing errors can be clearly implicated as causative mutations for a trait or disease. Insertions and deletions (indels) of nucleotides of a gene, both large and small, can alter the reading frame of a protein. They are found in genes such as agouti signaling protein (ASIP), which change a cat coloration from nonsolid to solid.7,8 A large sequence inversion clearly causes spinal muscular atrophy in the Maine Coon Cat.35 This sequence inversion is so large that it affects the end of one gene and the beginning of another, complicating the interpretation of which gene actually causes the disease.
Because of the different ways genetic mutations can potentially disrupt a protein, the validation of genetic tests can be a long and arduous process. SNPs that cause an amino acid in the protein to change, but not as drastically as causing a stop codon or splicing error (like both HCM mutations in cats), may affect some binding site or function of the protein. These mutations often are the most difficult to validate, because one must show cause and effect between the mutation and the disease. Computer modeling can support that the amino acid changes caused by the mutations would significantly alter the protein’s secondary and tertiary structures. Often a relative risk will be calculated between having the mutation and the disease in cases in which a cause and effect can not be defined clearly. For example, several DNA variants (SNPs) have been found in the cat HCM gene, myosin-binding protein C (MYBPC), but the relative risks associated with these additional mutations and having disease are lower than the mutations identified initially as causing Maine Coon and Ragdoll HCM.19,20
In addition, some confusion has arisen over “nomenclature” in regard to HCM. HCM in Maine Coons and Ragdolls is an autosomal dominant trait, implying only one copy of the mutation can cause the disease. Disease onset is unpredictable and some cats are severely affected and die young, while others have minimal cardiac involvement and live long lives. However, for most other dominant traits, when two copies are present and the mutation is homozygous, these individuals are severely compromised and die in utero (see Chapter 39). Taillessness in the Manx cat is a typical example. Breeding experiments suggested that the smaller litters associated with breeding two tailless cats are caused by homozygous lethality.36 The tailless mutation is still not known, but the mutations for different polydactyly in cats have been identified. Polydactyly is another autosomal dominant condition, but homozygous cats have been identified.37 In the case of PKD no homozygotes are found, even in severe and early-onset cases.11 But for HCM, cats homozygous for the Maine Coon mutation have been identified and usually have more severe disease with an earlier onset. Some investigators have erroneously considered the HCM mutation recessive because of the earlier age of onset in homozygotes. Because cats with one copy of the mutation can get disease, albeit generally at an older age, the disease still is an autosomal dominant trait, with variable age of onset and variable expression. These nomenclature disagreements can be very misleading, and can cause confusion and unfounded distrust in genetic testing.
Another reason for confusion and mistrust in genetic testing stems from situations in which more than one mutation in a gene can have equally plausible explanations for producing a trait. The recent identification of mutations associated with blood group B in the cat is a good example of the complications associated with defining causative mutations.38 Cytidine monophospho-N-acetylneuraminic acid hydroxylase (CMAH) is an enzyme that converts N-acetylneuraminic acid (NeuAc), the blood cell antigen associated with cat blood group A, to N-glycolylneuraminic acid (NeuGc), the antigen associated with cat blood group B. Several mutations have been identified in the CMAH gene, all completely associated with cat blood group B. Only one mutation should cause the B blood group; therefore why is there an association with several mutations in one gene? Sometimes extensive validation studies are required to elucidate the truly causative mutations for a trait. (See Chapter 61 for a discussion of feline blood groups.)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


