CHAPTER 56 Gene Therapy for Lysosomal Storage Diseases
Lysosomes and their enzymes degrade glycoproteins, polysaccharides, and complex lipids that have been taken into the cell by endocytosis or autophagy.1 Diseases resulting from defective lysosomal function and from the lysosomal accumulation of their substrates are known as lysosomal storage diseases (LSDs). In most cases a genetic deficiency of a specific lysosomal enzyme’s activity results in the accumulation of its specific substrate, causing cell swelling or cell death (Figure 56-1). In a small number of LSDs substrate storage occurs without defective lysosomal enzyme activity. In these cases a genetic deficiency of a structural protein, transporter protein, enzyme cofactor, or recognition marker is present, resulting in abnormal lysosomal function.
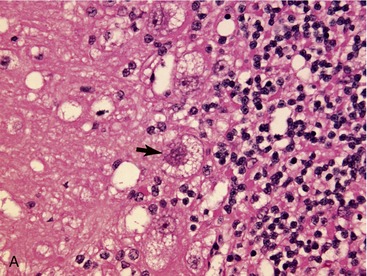
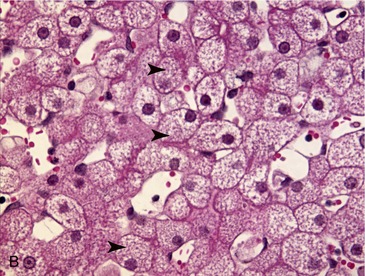
LSDs are inherited predominantly as autosomal recessive traits with the exception of two X-linked diseases: mucopolysaccharidosis II and Fabry disease.2 Abnormalities of multiple organ systems occur. Central nervous system (CNS) dysfunction and neuropathology are common. In the majority of cases swollen neurons and/or swollen glia can be identified histologically.3,4 Evidence of neurodegeneration (Figure 56-2), including cell loss, ubiquitin and neurofilament inclusions, and astrogliosis may be found.5,6 In some diseases toxic metabolites accumulate, resulting in large areas of necrosis.7 In LSDs with secondary changes in ganglioside metabolism, meganeurites, neurite sprouting, and abnormal synaptic connections develop.4,8–12 It remains unclear what effect cell swelling, neuronal degeneration and necrosis, secondary changes in gangliosides and other metabolites, and abnormal synaptic connections have on the development of the clinical signs of CNS dysfunction.
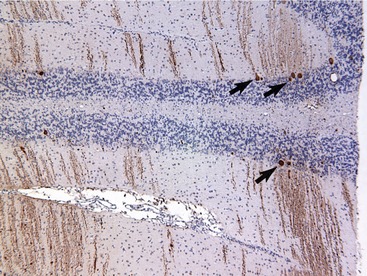
Over 45 different LSDs have been described in various species and no effective therapy exists to treat the majority of these diseases. A large number of murine and large animal models of LSDs have been identified with 12 described in cats.13–15 All cats described thus far have been under 1 year of age. A brief review of clinical signs associated with these diseases in the cat is provided in the following and a detailed review of the specific LSDs also is provided in Chapter 51 in the fourth volume of this series.
ALPHA-MANNOSIDOSIS
Alpha-mannosidosis (AMD) is caused by deficient activity of lysosomal α-mannosidase (LAMAN) that results in defective glycoprotein degradation and the intracellular accumulation of mannose-rich oligosaccharides.16,17 Disease in cats is characterized by progressive signs of cerebellar dysfunction, corneal and lenticular opacities, gingival hyperplasia, skeletal abnormalities, thymic aplasia, hepatomegaly, and polycystic kidneys (Figure 56-3).18–22 The cDNA encoding lysosomal α-mannosidase (MANB) has been cloned, and disease in Persian cats is caused by a four base pair deletion in the α-mannosidase gene resulting in a frameshift and premature termination.23 The mutation in affected domestic longhair cats and in domestic shorthair cats has not been determined.24,25 Neuropathological findings include vacuolated neurons, glial cells, and endothelial cells throughout the brain and spinal cord; Purkinje cell loss; and myelin deficiency of the CNS and peripheral nervous system (PNS).18,19,21,22,24 Meganeurite, neurite sprouting, and axonal spheroids also are described.26 Hepatocellular swelling occurs. The disease is diagnosed by assaying LAMAN activity in serum, white blood cells, or fibroblasts, or by testing for the known mutation.


NEURONAL CEROID LIPOFUSCINOSIS
Neuronal ceroid lipofuscinoses (NCL) are a group of at least eight distinct disorders (including Batten disease) caused by mutations in CLN genes (which produce both lysosomal enzymes and membrane-associated proteins) or the cathepsin D gene. The molecular and biochemical mechanisms leading to disease are not well understood.27 NCL has been described in Siamese cats, Japanese domestic cats, and domestic shorthair cats in Europe.28,29 Clinical signs included visual defects, ataxia, postural reaction deficits, myoclonus, and seizures in cats under 1 year of age. Brain atrophy, dilated cerebral ventricles, gliosis of the cerebral cortex and cerebellum, and loss of Purkinje cells are found. Cytoplasmic accumulation of autofluorescent material containing protein, lipid, and carbohydrate, occurs in the brain and other organs. Subunit C of mitochondrial ATP synthase is a major protein component of the stored material in most forms of NCL. The mutation resulting in NCL in cats has not been identified and diagnosis is made on the basis of signalment, clinical signs, electron microscopic evaluation of tissue, and—when present—assays of specific lysosomal enzyme deficiencies.
GALACTOSYLCERAMIDE LIPIDOSIS
Galactosylceramide lipidosis (globoid cell leukodystrophy [GLD]; Krabbe disease) is caused by mutations in the gene encoding galactosylceramidase (GALC) leading to accumulation of the toxic metabolite psychosine that results in destruction of oligodendroglia.30,31 GLD has been described in domestic shorthair kittens and a longhaired kitten.32–34 Clinical signs include dysmetria, generalized tremors, and paraplegia. Histological evaluation of the brain and spinal cord showed bilateral and symmetrical loss of myelin and axons, PAS-positive mononuclear cells (globoid cells), and astrogliosis. Globoid cells also may be found in the cerebrospinal fluid. Disease in cats has not been characterized enzymatically or genetically, and the diagnosis is made by clinical signs and measuring GALC activity in leukocytes or cultured fibroblasts.
GANGLIOSIDOSES
GM1 gangliosidosis is caused by a mutation in the gene encoding lysosomal acid beta-galactosidase resulting in the storage of the GM1 ganglioside and oligosaccharides. The disease has been described in Siamese cats, domestic shorthair cats, and Korat cats.35–39 Affected kittens developed generalized tremors, dysmetria, spastic quadriplegia, blindness, and seizures. Microscopic analysis of the nervous system shows vacuolization of neurons in brain, retina, spinal cord, and ganglia; cerebellar Purkinje cell and granular cell loss; the development of meganeurites and abnormal synaptic connections; and diminished myelination. Hepatocellular vacuolation also occurs. The disease is diagnosed by measuring decreased enzyme activity in leukocytes, or by identification of the recently identified mutation of the GLB1 gene.40
GM2 gangliosidosis is caused by mutations in the genes encoding beta-hexosaminidase, HEXA and HEXB, or in the gene encoding the cofactor necessary for hydrolysis of GM2 ganglioside (GM2A). Defects in any of these three genes may result in the storage of GM2 ganglioside and associated disease. Three forms of GM2 gangliosidoses are described in human patients: Tay-Sachs disease (mutation in HEXA), Sandhoff disease (mutation in HEXB), and GM2 activator deficiency (caused by a mutation in GM2A).41 GM2 gangliosidosis is homologous to Sandhoff disease and has been described in shorthaired cats and Korat cats.42–46 Clinical signs in affected cats include corneal clouding, hypermetria, head tremors, seizures, and paralysis. Histological examination shows vacuolization of neurons and hepatocytes, and the development of meganeurites and abnormal synaptic connections.47 Diagnosis is made by confirming decreased enzyme activity, or by identification of mutations that have been identified in both cat breeds.45,48
GLYCOGENOSES
Glycogen storage disease type II (acid maltase deficiency; Pompe disease) is caused by a mutation in the gene encoding acid alpha-glucosidase leading to the accumulation of glycogen in skeletal and cardiac muscle, as well as in other tissue.49 Glycogen storage disease type II has been described in one cat.50 No clinical abnormalities were described in this report. Histological evaluations showed cytoplasmic storage of glycogen in spinal cord neurons (the brain was not examined). Disease in cats has not been characterized enzymatically or genetically.
MUCOLIPIDOSIS
Mucolipidosis II (MLII; I-Cell disease) is caused by a defect in N-acetylglucosamine-1-phosphotransferase (GNPTA), which is responsible for the posttranslational addition of the mannose 6-phosphate recognition marker to lysosomal enzymes, a necessity for targeting the lysosomal enzyme to the lysosome. In MLII, lysosomal enzymes are not internalized in the lysosome and instead are secreted into the extracellular space. Affected cats show mental dullness, facial dysmorphia, retinal degeneration, skeletal abnormalities, paraparesis, and ataxia.51–53 Light microscopic changes include widespread storage of material in fibroblasts, endothelial cells, cartilage, and heart valves. A missense point mutation in the GNPTA gene has been identified that results in a stop codon leading to premature termination of the coding sequence.54
MUCOPOLYSACCHARIDOSES
The mucopolysaccharidoses (MPS) are a group of lysosomal storage diseases caused by deficiencies of enzymes involved in the degradation of glycosaminoglycans (mucopolysaccharides). Multisystemic disease with or without nervous system dysfunction is described. Preliminary diagnosis of MPS disease is made by finding increased urinary excretion of glycosaminoglycans, and confirmed by finding decreased serum or tissue concentrations of the individual lysosomal enzymes.55
MPS I (Hurler, Scheie, and Hurler/Scheie syndromes) is caused by alpha-L-iduronidase (IDUA) deficiency resulting in storage of dermatan and heparan sulfates. MPS I cats show growth retardation, dysostosis multiplex, facial deformity, lameness, corneal opacity, and cardiac murmurs (Figure 56-4).56–58 No measurable neurological dysfunction is present. Pathology includes multiple epiphyseal dysplasia, mitral valve thickening, and cytoplasmic vacuolization of neurons, hepatocytes, and other cell types. The diagnosis is made by finding low enzyme activity in leukocytes, or confirming the presence of the causative mutation, a three base pair deletion in the IDUA gene.59
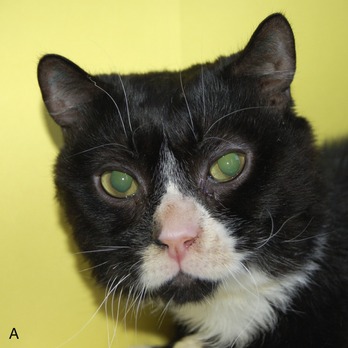
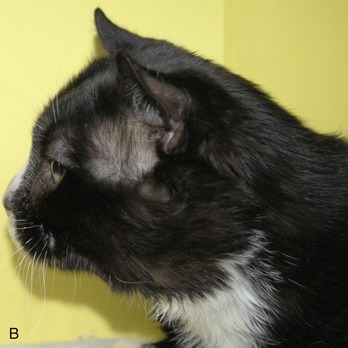
MPS VI (Maroteaux-Lamy syndrome) is caused by deficient activity of N-acetylgalactosamine 4-sulfatase (arylsulfatase B) resulting in the storage of dermatan sulfate. Affected cats are dwarfed with facial deformity, corneal opacity, joint laxity and swelling, lameness, and fusion of the cervical and lumbar spine (Figure 56-5).60–64 Histological findings include epiphyseal dysplasia and cytoplasmic vacuolization of many tissues. The CNS is not involved. The causative mutation is a point mutation in the arylsulfatase B gene.
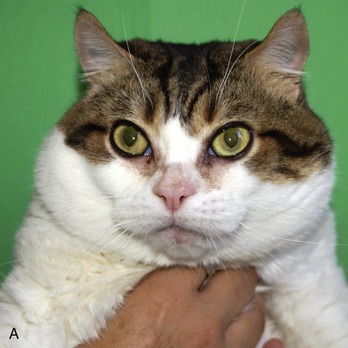
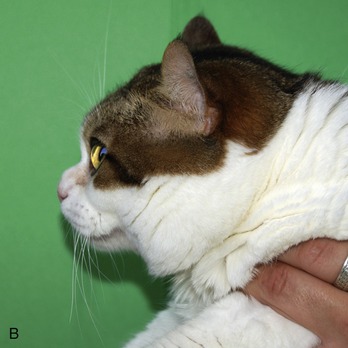
MPS VII (Sly syndrome) is caused by deficient beta-glucuronidase (GUSB) activity resulting in the storage of dermatan sulfate, heparan sulfate, and chondroitin 4-, 6-sulfates.55 Clinical signs include growth retardation, facial and thoracic deformity, corneal opacity, joint swelling, lameness, and cardiac murmurs.65 The gene encoding GUSB has been cloned in cats and the point mutation identified.66
SPHINGOMYELINOSES
Sphingomyelinoses types A and B (Niemann-Pick disease type A and B) are both caused by deficient activity of acid sphingomyelinase and the accumulation of sphingomyelin.67 Affected Siamese cats with Niemann-Pick type A show tremors, ataxia, and pelvic limb weakness. Histopathological examination shows vacuolization of neurons, hepatocytes, and cells of the reticuloendothelial system.68 Molecular characterization of the disease in cats has not been performed.
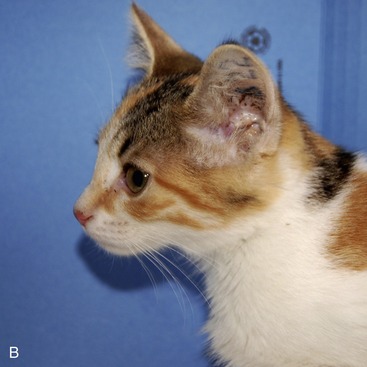
Niemann-Pick type C (sphingomyelinosis type C) is caused by a mutation in either the NPC1 or NPC2 gene.69,70 The proteins NPC1 and NPC2 are critical for the movement of unesterified cholesterol and glycosphingolipids from the endosomal/lysosomal compartment to the Golgi apparatus, plasma membrane, and endoplasmic reticulum. NPC1 is a membrane-bound protein and NPC2 a soluble protein. Mutations in either gene result in the lysosomal accumulation of unesterified cholesterol and glycosphingolipids. Cats with NPC disease have a spontaneously-occurring missense mutation in NPC1 and show progressive signs of cerebellar dysfunction and hepatic disease (Figure 56-6).71–76 Histological evaluation shows Purkinje cell loss and storage of lipid in neurons, hepatocytes, and macrophages; ectopic dendritogenesis and axonal spheroid formation also occur.77 Diagnosis is made by molecular testing for the known mutation, by identification of the intralysosomal storage of unesterified cholesterol, and/or biochemical evidence of impaired LDL-induced cholesterol esterification.

THERAPY OF LYSOSOMAL STORAGE DISEASES
Lysosomal hydrolases are synthesized as pre-proenzymes on endoplasmic reticulum–bound ribosomes, and modified to their functional form in the endoplasmic reticulum and Golgi apparatus. In the Golgi apparatus a mannose 6-phosphate (M6P) recognition marker is added, which is recognized by M6P receptors on the Golgi membrane, allowing transfer of the enzyme to the prelysosomal/endosomal vesicles. The low pH within these vesicles results in dissociation of the lysosomal enzyme from the receptor. The receptor recycles to the Golgi apparatus and the lysosomal enzyme is packaged into lysosomes.78,79 A portion of the M6P-modified enzyme also is released into the extracellular space and is responsible for the lysosomal enzyme activity found in the extracellular space and in serum. Secreted enzyme may be endocytosed by cells expressing M6P receptors on their cell membrane. Intracellularly, the lysosomal enzyme dissociates from the receptor and is packaged into lysosomes.79–81
Therapeutic approaches for LSDs are based on this knowledge that lysosomal enzymes are secreted and endocytosed by neighboring cells, and can be transferred by direct cell-to-cell exchange.82–84 Therefore the goal of therapy has been to deliver functioning M6P-conjugated enzyme to the enzyme-deficient patient in whom receptor-mediated uptake of the normal lysosomal enzyme by diseased cells will result in correction of pathology. Three general treatment strategies have been developed: enzyme replacement therapy, heterologous cell or organ transplantation, and somatic gene transfer.
ENZYME REPLACEMENT THERAPY
Enzyme replacement therapy with recombinant glucocerebrosidase injections is a clinical success in the major form of human Gaucher disease, an LSD without CNS involvement.85 However, in LSDs in which signs of CNS dysfunction are the predominant feature, intravenously administered enzyme appears incapable of crossing the blood-brain barrier in concentrations necessary to sufficiently treat the brain.86 The drawbacks for enzyme replacement therapy include the need to produce the recombinant protein, the need for lifelong therapy, high cost, the development of antibodies to the enzyme, and incomplete response to therapy.
Enzyme replacement therapy with N-acetylgalactosamine-4-sulfatase has been used in the therapy of feline MPS VI and MPS I.87–91 Affected MPS VI cats treated weekly from birth showed a reduction in urinary glycosaminoglycan concentrations and improvement in lysosomal storage in heart, aorta, skin, dura, liver, and perivascular cells of the brain. Therapy also resulted in improvement of bone pathology, improved mobility of the cervical spine, and less spinal cord compression. Treatment of six MPS I cats resulted in diminished glycosaminoglycan storage in liver and spleen and improvement in histological abnormalities. There was no improvement in CNS pathology.
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


