Chapter 19 Gastrointestinal Pharmacology
Drugs that Target Appetite or Caloric Balance
Appetite is controlled primarily, but not exclusively, by the ventral and lateral nuclei of the hypothalamus. The nuclei respond to both short- and long-term signals.1 Hypothalamic directives are influenced by energy, which in turn is influenced by ingestion, absorption, metabolism, and storage. Chemical signals mediating these directives act locally or distantly, often balancing one another. Signals include hormones, neuropeptides, cytokines and neurotransmitters, several of which are pharmacologic targets.
Although a discussion of energy utilization is too complex and extensive for this text, understanding of the role of body fat in appetitic control and body weight has been markedly advanced in the last decade and warrants review of those aspects relevant to drug therapy. In the arcuate nucleus, primary neurons detect concentrations of metabolites and secondary neurons synchronize signals and coordinate vagally mediated responses. Among these neurons are two distinct populations of primary neurons and neuropeptides that control food intake and energy expenditure. Orexigenic peptides increase food intake and include neuropeptide Y (NPY) and agouti-related protein (AgRP); gamma amino butyric acid (GABA) is also released as an orexigenic signal. The NPY/AgRP neurons direct the effects of leptin and ghrelin and stimulate feeding during states of fasting. They also influence the anorexigenic peptides, which decrease food intake. Anorexigenic peptids include the pro-opiomelanocortin (POMC) neurons which produce alpha-melanocyte hormone (α-MSH), a powerful appetite suppressant, and the β-endorphins.. The POMC extensively communicates with hypothalamic neurons as well as other regions that regulate energy. Both orexigenic and anorexigenic neurons have receptors for balancing signaling molecules, which often cause opposing effects by interacting at the same ligand. NPY and α-MSH, located in the ventromedial nucleus of the appetite center, are considered balancing hormones. Other stimulatory mediators include norepinephrine (α2-receptors), dopamine (possibly D1 receptors), and opiate and pancreatic polypeptides. Although GABA also stimulates appetite, its effect may vary with route of administration.1 Other inhibitory mediators include serotonin (5-hydroxytryptamine [5-HT]), calcitonin, cholecystokinin, and corticotrophin-releasing factor. In addition to their central roles, several of these mediators influence energy metabolism peripherally.
Among the major target tissues for signals modulating energy metabolism is adipose tissue, which is now a recognized endocrine organ (Figure 19-1). Hormones play a critical role in energy homeostasis, insulin sensitivity, and lipid and carbohydrate metabolism. The system appears to be more sensitive to preventing starvation rather than obesity. Leptin and insulin appear to be the predominant mediators that signal adiposity: both circulate in proportion to body fat, enter the central nervous system (CNS) in proportion to plasma concentrations, and interact with CNS receptors that influence energy regulation, as reviewed by Havel.2 Other important peripheral mediators include acetylation-stimulating protein (ASP) and adiponectin, each being principally regulated by host nutritional status.
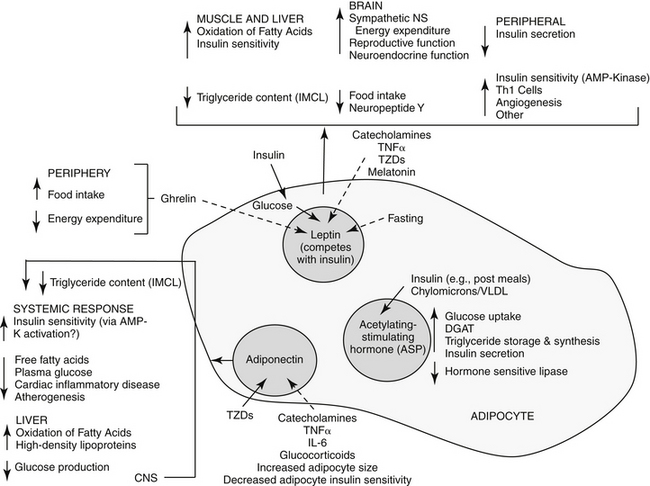
Leptin was discovered in obese mice that randomly emerged as mutants among normal populations. At least six leptin receptors (“LepR”, a through f) are associated with the ob gene (Ob-R) (obesity gene), suggesting a complex role for this hormone. 3 Leptin receptors are similar to class I cytokine receptors, perhaps linking cytokines to diseases associated with obesity disorders.3
Currently, little information appears to be available regarding leptin content in dogs or cats and information must be drawn from other species. Although most organs produce leptin, white adipose tissue is the primary source, with the amount produced varying among species: more leptin is produced in subcutaneous compared with omental fat in humans, but the opposite is true in rats.3 In humans, leptin content is greatest in the heart, liver, small intestine, prostate, and ovaries versus the lung and kidney in mice. Each tissue may have a particular receptor isoform, allowing differential roles among tissues. Leptin plays an important, albeit complex, role in the peripheral regulation of adipose tissues. Its primary function is as a lipostat, communicating to other tissues the current status of body fat reserves. As such, leptin mediates fuel movement and use (see Figure 19-1) and energy expenditure. 3 It is more effective in signaling deficient rather than excessive energy reserves.2 Activated leptin receptors induce satiety in the appetite center (inhibition of NPY, stimulation of alpha-MSH neurons). Leptin also appears to influence thermogenesis. Peripherally, leptin appears to increase hematopoiesis, angiogenesis, wound repair, and puberty; its role in reproduction is particularly complex. Leptin is primarily regulated by food-induced responses to insulin, with influences being more dramatic during fasting and characterized by diurnal variation.2 In normal animals, insulin appears to directly regulate leptin gene expression and excretion in adipose tissue; it also indirectly influences it through adipocyte glucose use and oxidative metabolism. Both hormones increase in concert.3 Both leptin and insulin share inhibitory signal transduction pathways in response to food.2 Leptin, in turn, appears to inhibit insulin secretion, and, peripherally competes with insulin; however conflicting results also suggest that leptin has no effect on insulin.2,3 Glucocorticoids appear to stimulate leptin gene expression, although it is not clear if this effect is direct (gene expression) or indirect (altered food intake or insulin concentrations). Leptin is downregulated by melatonin at night; accordingly, humans with short sleep cycles may be predisposed to obesity. Depletion of body stores is not detected in hypoleptimemic animals, and as such, is more dangerous than hyperleptinemia. In humans, hypoleptinemia is associated with neuroendocrine, reproductive, metabolic, and immunologic imbalances,and in rodents, marked insulin reistance and hyperlipidemia.2 Leptin concentrations in persons with eating disorders characterized by anorexia are similar to or lower than concentrations in persons without eating disorders.3. Its role in chronic liver disease increasingly is being recognized.4 Many of these abnormalities can be normalized in humans with recombinant human leptin. Differences in genes (including mutations) regulating leptin also are linked to human obesity with hyperleptinemia indicating leptin resistance.
KEY POINT 19-3
Adiponectin influences lipid and carbohydrate metabolism and is necessary for normal insulin action.
The second major hormone influencing energy metabolism, ASP, is produced from complement factor C. Locally, ASP paracrine actions in adipose tissue include increased glucose uptake and diacylglycerol acyltransferase activity and decreased hormone-sensitive lipase (see Figure 19-1). As such, adipocyte triglyceride synthesis and storage increase after eating, resulting in increased free fatty acids and triglyceride clearance. Serum ASP concentrations are increased by lipids, and increase in proportion to body fat in obese human patients; they decrease during states of fasting. Insulin may control ASP in reponse to eating or fasting; ASP in turn may directly stimulate insulin secretion.
Adiponectin, a large protein secreted by adiopocytes, is a third hormone influencing energy metabolism. Its biological effect varies with its state of diamerization.2 Adiponectin appears to influence lipid and carbohydrate metabolism both directly and indirectly (see Figure 19-1). Adiponectin appears to be necessary for normal insulin actions. Concentrations are reduced in patients with type 2 diabetes compared with nondiabetic humans.2 The effect of adiponectin on decreased circulating glucose are independent of fat content and occurs without influencing insulin secretion. Mechanisms may include decreased hepatic glucose formation and increased tissue glucose use by decreasing insulin resistance. Circulating adiponectin is negatively correlated with the content of body fat, particularly visceral (e.g., omental) rather than subcutaneous in humans.2 Adiponectin may reduce ectopic fat in the liver and muscle through increased fat oxidation. Thus, low visceral fat may reduce adiponectin production, contributing to insulin resistance associated with obesity.2 The impact of insulin on adiponectin is not clear. Among the emerging effects of adiponectin is protection against inflammatory cardiovascular disease (including atherosclerosis), which may explains the relationship between body fat and cardiac disease. Cytokines, catecholamines, and glucocorticoids decrease adiponectin production, which may contribute to their characteristic incease in insulin resistance.
Appetite Stimulants
Studies regarding the pharmacologic control of appetite have traditionally focused on decreased, rather than increased, food intake, particularly in humans. However, the role of cachexia associated with weight loss and anorexia in human patients with cancer has stimulated a renewed interest in appetite stimulants.5 Drugs that inhibit gluconeogenesis, such as hydrazine sulfate, or promote gastric emptying, such as metoclopramide, have been used successfully to stimulate food intake in some human patients.5 The use of steroids, including megestrol acetate, in treatment of cachexia is discussed later in this chapter.5 Both glucocorticoids and B vitamins have been used to nonspecifically stimulate appetite in animals. Drugs used to treat depression and psychosis in human patients are associated with appetite increase and weight gain.6 They antagonize a variety of receptors, although their clinical potency is often related to increased serotonin, which may, in fact, decrease appetite in some patients.
Mirtazapine is a piperazino-azepine antidepressant characterized by serotonergic activity as a result of 5-HT-1 agonistic activity and inhibition of serotonin reuptake.7 Sympathetic (norepinephrine) actions reflect antagonism of α –2 autoreceptors as well as influences by other receptors. As a behavior-modifying drug, mirtazapine is discussed in greater depth in Chapter 26. Anecdotally, mirtazapine has been used to stimulate appetite in either dogs or cats, the latter at 3 mg/cat every 72 hours.
The benzodiazepines diazepam (Valium) and oxazepam, a metabolite of diazepam (Serax), have successfully induced appetite in cats, probably through gabaminergic effects and central inhibition of the satiety center in the hypothalamus (Table 19-1).8 Diazepam is administered intravenously or orally, whereas oxazepam is administered orally. Of the two drugs, diazepam may be more effective, although sedation is greater. The benzodiazepines do not stimulate appetite in the dog as effectively as in the cat. Hepatotoxicity associated with diazepam therapy when used as an appetite stimulant has been reported in cats9,10 and is discussed in more depth in Chapter 27. Toxicity appears to be idiosyncratic and thus may not be predictable; it is not likely to happen in a large percentage of animals receiving the drug.
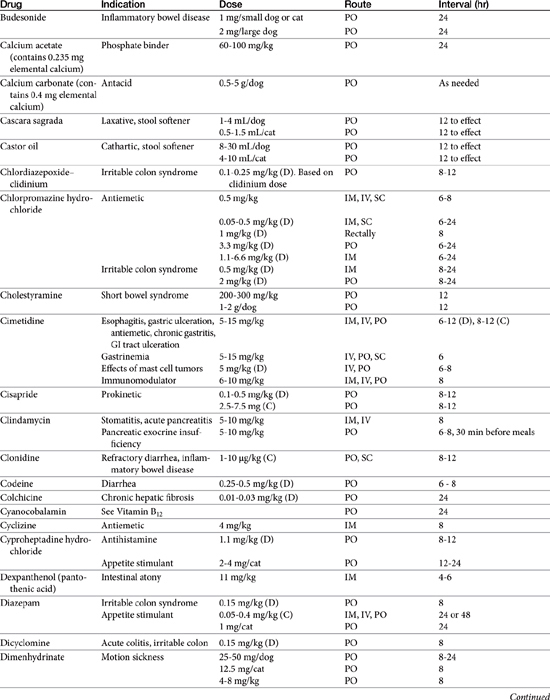
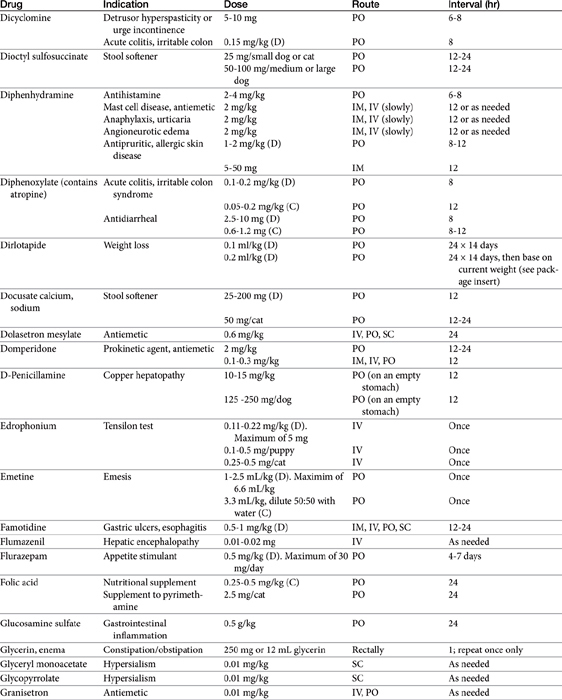
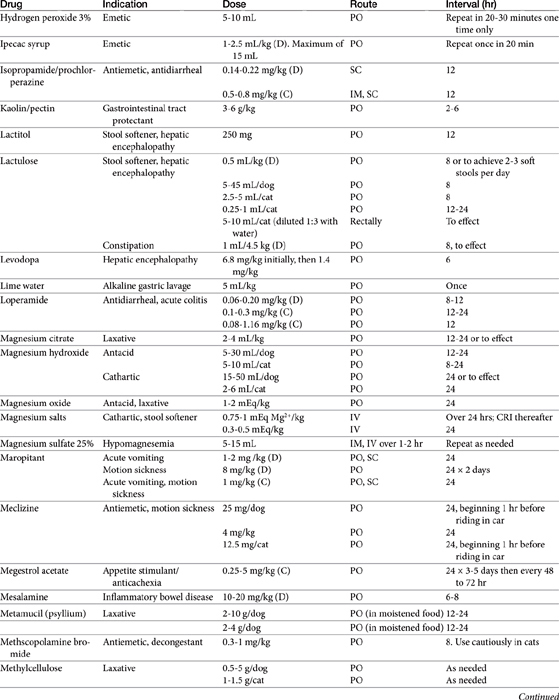
Cyproheptadine, an antihistamine with antiserotonin properties, has caused weight gain in geriatric human patients and in adults and younger patients afflicted with eating disorders. Its mechanism probably reflects inhibition of serotonergic receptors that control appetite. Serotonin antagonists also increase food intake in cats,1 and cyproheptadine has been used clinically to stimulate the appetite of some anorexic cats. Cyproheptadine kinetics have been reported in the cat. Oral bioavailability of the tablet is 100%, and the elimination half-life approximates 13 hours.11 Cats tolerated a dose of 8 mg orally with no adverse effects, although impact on appetite was not described. Based on this study, once- to twice-daily dosing of 8 mg appears to be safe.
Cachexia
Cachexia is an involuntary state characterized by loss of more than 5% of body weight. In humans it occurs over a defined period, generally of 2 to 6 months. Ultimately, it is a condition of starvation characterized by depletion of body mass, particularly muscle, but to a lesser degree, adipose tissue.12 Cachexia develops in 50% of human patients with cancer, contributing to substantially shortened survival times. Cachexia also is a manifestation of other chronic diseases, including (in humans), acquired immune-deficiency syndrome (AIDS), heart failure, rheumatoid arthritis, Crohn’s disease, chronic obstructive pulmonary disease, and chronic renal disease.12 It appears to be a cytokine-driven process, with key components including anorexia and a state of hypercatabolism. The principle cytokines appear to include TNF-alpha, interleukins 1 and 6, and interferon-γ Of these, TNF-alpha is presumed to stimulate mechanisms that lead to severe cachexia.12 Muscle wasting may reflect inhibition of myogenic differentiation; an energy sink may result from increased concentrations of mitochondrial uncoupling proteins. Lipoproteinase activity is inhibited by TNF-alpha. Interleukins contribute to CNS-mediated anorexia and decreased albumin synthesis in the liver. Megestrol acetate is the only treatment approved by the Food and Drug Administration for cancer or AIDS-related cachexia syndromes in humans.12 Other less commonly used drugs are glucocorticoids, anabolic steroids, antiseratonergic drugs, dronabinol, and prokinetic drugs.
Megestrol is a synthetic derivative of progesterone.12 As such, megestrol acetate targets cachexia by directly and indirectly stimulating appetite and antagonizing the catabolic metabolic effects of cytokines. As with other steroidal hormones, the effects of megestrol (and progesterone) involve passive diffusion into the cell and binding to specific intracellular progesterone receptors A or B (PR-A or -B) and heat shock proteins. The drugs move into the nucleus, bind to progesterone response elements on target genes, and influence transcription through inhibition (PR-A) or stimulation (PR-B) of other steroid response elements. Although the majority of progesterone effects are genomic, nongenomic actions also occur. Metabolic effects of progesterone include increased basal insulin concentration and increased response to carbohydrate load; increased lipoprotein lipase with altered fat deposition, plasma lipid, and lipoprotein concentrations; and modulation of body temperature.12 In humans and animal models of cancer, megestrol stimulates appetite, increases caloric intake, induces a sense of well-being, and causes weight gain, particularly of fat. Fat is the preferred weight gain because it provides more kilocalories per gram than proteins or carbohydrate and helps stabilize body temperatures.12 Megestrol decreases the effects, sometimes by inhibiting formation of TNF-alpha, interleukin-1 (IL-1), and interleukin-6 (IL-6). Centrally, megestrol appears to modulate neurotransmitters responsible for appetite regulation such as NPY, which in turn stimulates the release of other mediators. Megestrol may also stabilize declining concentrations of β-endorphins in the cerebrospinal fluid.
Effects of megestrol are dose and (particularly for fat gain) duration dependent. Initial weight gain requires high concentrations (>300 ng/mL) for more than 40% of a 24-hour dosing interval. In humans this requires administration of the tablet four times daily. Megestrol acetate is used rather than other progestationals, which must be given parenterally.12 Bioavailabilty of megestrol acetate is variable, being greater with the oral solution compared to the tablet. With tablets, peak concentrations may vary 6 fold; variability is much less with the oral solution. Disposition is complex. Hepatic metabolism is necessary to free the steroid from acetate, with the steroids subsequently conjugated with glucuronic acid before elimination. Elimination also varies, with half-lives that range from 13 to 105 hours. In humans, a single daily dose of the suspension (800 mg) achieves peak concentrations between 1500 and 3000 ng/mL. Not surprisingly, the oral solution is associated with a much higher rate of response compared with the tablet. A micronized (nanocrystal) preparation is currently under investigation for human use.
Limited information is available regarding use of megestrol acetate in animals. The disposition of megestrol acetate has been described in Beagles as part of the preclinical assessment in humans. Four preparations were studied for 72 hours after administration of 10 mg/kg (by oral gavage) either in the fasted or the fed (high-fat meal) state. The preparations included two different nanocrystal oral solutions and two commercially available oral suspensions (Par Pharmaceutical and Bristol-Myers). After the high-fat meal, peak concentrations (1600 to 2200 ng/mL) and area under the curve (AUC) were higher with the nanocrystal oral suspensions compared with the commercially available oral suspensions (both approximating 300 ng/mL). Although the elimination half-life was not reported, the disappearance half-life of megestrol appeared to be between 10 and 20 hours.12
Megestrol acetate appears to be better tolerated in humans compared with animals. The primary adverse events in humans are thromobembolic, reflecting increased thrombin receptors in smooth muscles. Venous distention and capacitance increase, contributing to reduced blood flow and stasis. Addison’s disease and glucose intolerance are sporadically reported, reflecting its intrinsic corticosteroid activities.12
Anecdotally, megesterol acetate has been effective in dogs to treat chemotherapy-induced nausea and inappetence. However, in humans a prospective randomized controlled clinical trial in cachectic human cancer patients compared the efficacy of an anabolic steroid (fluoxymesterone [10 mg, 0.142 mg/k] twice daily), megestrol acetate tablets (800 mg [11.4 mg/kg] once daily), and a glucocorticoid (dexamethasone, 0.75 mg [0.01 mg/kg] four times daily) as appetite stimulants. Of the three, the anabolic steroid was least (significantly) and megestrol (nonsignificantly) most clinically effective. Glucocorticoids usually were discontinued because of side effects, although megestrol acetate was associated with the most thromboembolic events.13
Appetite Suppressants and Anti-Obesity Drugs
Obesity is a physiologic disorder of energy balance in which energy intake exceeds energy expenditures. Excessive energy is stored as fat. In rodent models and humans, leptin deficiency or leptin resistance can result in obesity caused by hyperphagia and decreased energy expenditure.3 Other characteristics of obesity in humans include non–insulin-dependent diabetes mellitus, severe insulin resistance, hypothermia and cold intolerance, infertility, and decreased lean body mass.3
Among the drugs studied for central suppressant effects are cannabinoids, the manipulation of which may influence consumption of highly palatable foods. For example, rimonabant is a cannabinoid receptor type 1 (CB1) receptor antagonist that decreases intake of palatable foods, leading to decreased body weight in rodents. However, dogs (and other species) respond differently to cannabinoids. For example, although CB1 antagonists decrease food intake, causing weight loss in dogs, appetite suppression is attenuated after several weeks. Dogs also appear more sensitive to side effects associated with CB1 antagonists, exhibiting vomiting, diarrhea, and pruritus at doses necessary to decrease appetite. Although cats tolerate antagonists better than dogs, they respond only at doses associated with severe pruritus, panting, agitation, and CNS stimulation.14 Selective 5-HT2C receptor antagonists appear to be effective in decreasing food intake in rodent models but not in dogs or cats. Higher doses that might be more effective are associated with adverse effects.14 The human pancreatic lipase inhibitor orlistat is associated with modest weight loss in dogs. However, significant increase in food intake, presumably in response to caloric loss, is accompanied by markedly increased fecal fat, leading to uncontrolled leakage and perianal and abdominal soiling.14
Use of dilortapide in dogs must be accompanied by a weight loss program. Loss of appetite will not last more than several days after therapy is discontinued. Dosing is complex and is based on body weight (see package insert). A total of 11% to 13% of body weight was lost in patients studied during drug approval (PI), an amount considered to contribute positively to animal health. At study end mean final dose was 0.26 to 0.56 mg/kg. Dilortapide should not be administered to humans or cats. Adverse reactions in humans include abdominal pain, distention, diarrhea, flatulence, nausea, and vomiting.
Several drugs may affect appetite secondary to their intended therapeutic effect. Propofol (1-2 mg/kg IV) was reported in a research abstract15 to be an effective appetite stimulant, presumably through stimulation of GABA-A and NYP and inhibition of serotonin receptors. Anecdotally, omega-3 fatty acids (EPA and DHA) may stimulate appetite by inhibiting cytokines responsible for anorexia.
Emetics and Antiemetics
The Vomiting Reflex
Emesis is a complex protective reflex that is not well developed in all species but does occur in both dogs and cats.16–18 Although several afferent pathways may be responsible for initiating emesis, all signals are coordinated by the emetic center. Located in the lateral reticular formation in the mid brainstem, the emetic center is in close proximity to the nucleus tractus solitarius of the vagus nerve and the chemoreceptor trigger zone (CTZ), the latter of which is located adjacent to the area postrema in the bottom of the lateral ventricle. The latter coordinates vomiting associated with blood-borne chemicals (Figures 19-2 and 19-3). The emetic center coordinates vomiting associated with afferent peripheral and central (neural) signals. Among the signals coordinating vomiting is the tachykinin neuropeptid, ≥substance P. Drugs that cause or ameliorate vomiting generally do so by modifying afferent or efferent neurotransmitters responsible for transmission of the signal from various afferent sites. The emetic center is protected by the blood–brain barrier, whereas the CTZ is not; therefore, the CTZ is able to monitor the presence of emetics in the blood or cerebrospinal fluid. However, drug penetrability to each site varies, affecting both drug safety and efficacy.16–20
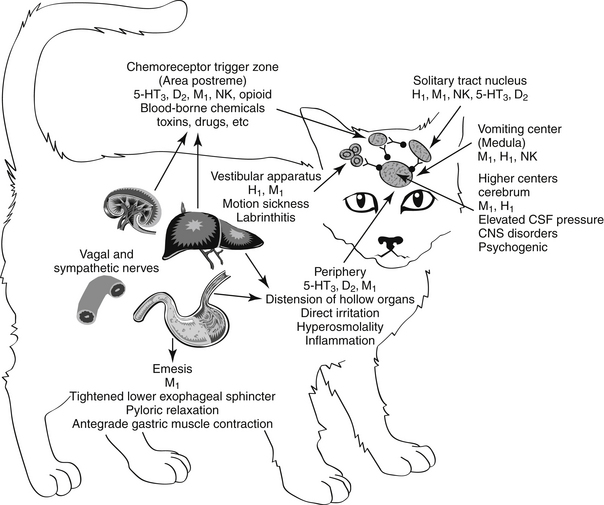
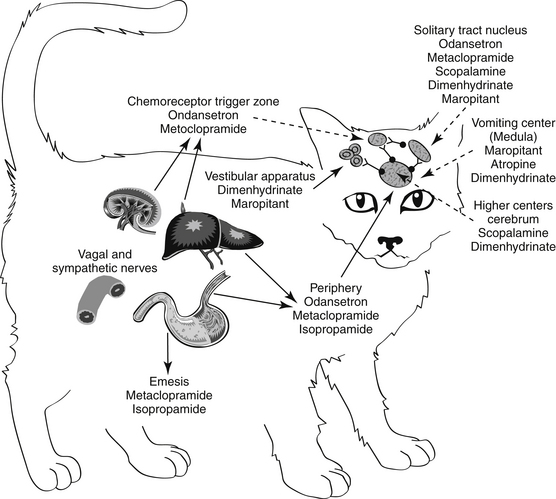
Figure 19-3 Antiemetic drugs effective at each site of the emetic reflex. CTZ, Chemoreceptor trigger zone.
Several sites in the vomiting reflex are targeted by drugs. Ideally, drugs that target the emetic center would be characterized by the broadest spectrum and potentially the best efficacy. Historically, because such drugs must be able to penetrate the blood–brain barrier, they tend to be characterized by increased risk of side effects. This risk is minimized if the drug targets a mediator whose effects are limited to the vomiting center only. In addition to integration of the emetic reflex, impulses integrated by the center include afferent signals from higher centers such as the cerebral cortex and limbic system. For example, psychogenic vomiting, or vomiting induced by visual and olfactory stimuli, originates in the cerebral cortex, whereas head injuries and increased intracranial pressure initiate emesis by way of the limbic pathways. The solitarius nucleus contains receptors for enkephalin, histamine, serotonin (5-HT3), and acetylcholine (ACh). ACh is a major afferent neurotransmitter in the higher centers, with histamine acting as a secondary transmitter by way of H1 receptors.19 However, substance P, a member of the tachykinin family of neuropeptides, has recently been identified as a key neurotransmitter associated with emesis in higher centers, including the emetic center.21 Substance P targets neurokinin (NK1) receptors located throughout the emetic center (as reviewed by Wu20), including the nucleus tractus solitarius, the area postrema, and the dorsal motor nucleus of the vagus.22 The emetic center also involves cannabinoid receptors (also located in the CTZ), although their role is not clear.
Blood-borne chemical compounds stimulate the CTZ.17,19 Examples include circulating toxins associated with disease (uremia, pyometra, liver disease, endotoxemia), radiation sickness, and drugs (e.g., opioids, cardiac glycosides, anticancer chemotherapeutic agents). Signals in the CTZ are mediated by dopaminergic (D2)19 and serotonergic (5-hydroxytryptamine; 5-HT3) receptors23 and neurokinin receptors. Histamine by way of H1 receptors acts as a secondary neurotransmitter at the CTZ. Neurokinin receptors in humans are responsible for the delayed phase of vomiting associated with cisplatin anticancer chemotherapy. Alpha-2 receptors associated with the area postrema also induce emesis in dogs, cats, and other species.24–26 The CTZ also is rich in opioid receptors. The safety of CTZ-active drugs may be increased by selectively targeting the subreceptor types for each neurotransmitter.
Emetic impulses originating from the semicircular canals of the vestibular apparatus are transmitted by the eighth cranial nerve to the vestibular nuclei and then by way of the CTZ and the uvula and nodulus of the cerebellum to the emetic center. This pathway, mediated by histaminergic (subtype H1) receptors, is responsible for eliciting the emesis that accompanies motion sickness and labyrinthitis.27
Peripheral impulses cause emesis that arises from stimulation of the pharynx and fauces; the signals are transmitted by afferent nerves in the ninth cranial nerve to the emetic center. Other peripheral afferent pathways include those arising from stimulation (i.e., irritation or distention) of various visceral organs and tissues. Impulses may be carried by sympathetic or vagal afferents from the heart, stomach, duodenum, small intestine, liver, gallbladder, peritoneum, kidneys, ureter, urinary bladder, and uterus. ACh is the primary neurotransmitter mediating the afferent limb of the emesis reflex from peripheral causes. Muscarinic receptors initiate the impulse that travels to the emetic center by way of the vagus nerve.
Efferent signals that stimulate the emetic reflex travel back to the stomach by the tenth cranial (vagus) nerve. ACh also acts as the primary efferent neurotransmitter in the vagus and in the smooth muscle of the stomach. In the stomach, dopamine receptors (D2) appear to inhibit gastric motility, during nausea and vomiting. In addition, dopamine receptors contribute to reflexes that allow relaxation of the upper stomach and delayed gastric emptying associated with gastric distention caused by food.24 Finally, serotonin (by way of 5-HT3 receptors) contributes afferent pathways from the stomach and small intestine.24
Emetics
Centrally Acting Emetics
Xylazine is an α2-agonist historically used for sedative analgesia. Emesis in dogs is not as consistent as in cats. Emesis mediated by α2 stimulation occurs in cats at doses lower than that recommended for sedation (0.05 mg/kg).26 Emetogens were evaluated in cats in anticipation of an antiemetic clinical trial.22 Three emetogens were tested, with xylazine (0.44 mg/kg intramuscularly) reliably causing emesis. In contrast, neither apomorphine (0.04 mg/kg intravenously) nor syrup of ipecac (0.5 mL/kg) predictably caused emesis. Syrup of ipecac causes anorexia for several days. The use of medetomidine to induce emesis has not been reported, although its actions are similar to those of xylazine.
Antiemetics
Antiemetics control emesis by either a central or a peripheral action (see Figures 19-2 and 19-3). Both actions depend on and can be correlated with blockade of neurotransmission at receptor sites.27,28 Centrally acting antiemetics block impulses at higher centers and at the emetic center and include muscarinic anticholinergics and drugs that target neurokinin receptors; antidopaminergics and antiserotonergics, which block dopaminergic receptors at the CTZ; and antihistaminergics, which primarily block H1 receptors at the vestibular apparatus but secondarily at multiple central centers. Antiemetic agents possess either a limited or a broad effect, depending on which signals and centers are inhibited.
Centrally Acting Antiemetics
Vomiting center
Maropitant (Cerenia) is a neurokinin (NK1) receptor antagonist that blocks the actions of substance P in the area postrema and nucleus solatarius; Aprepitant is a human drug in the same classused as rescue anti-emetic therapy in cancer patients nonresponsive to 5HT3–dexamethasone combinations.29 Approved as an oral or subcutaneous preparation for dogs for the prevention of acute vomiting and motion sickness, maropitant has proved efficacious in the control of vomiting associated with many central and peripheral causes (Table 19-2).
In dogs bioavailability is greater after subcutaneous (91%) compared with oral (24%) administration, probably because of first-pass metabolism (PI). Relevant pharmacokinetic parameters include Cmax (ng/mL) of 92 and 81 ng/mL at 0.75 and 2 hours, respectively, after administration of 1 mg/kg subcutaneously and 2 mg/kg orally, respectively. It is highly (99.5%) bound to plasma proteins. Maropitant is metabolized in dogs by CYP2D15 and CYP3A12. Elimination half-life in dogs approximates 9 hours after subcutaneous administration and 4 to 5 hours after oral administration. However, saturation of drug-metabolizing enzymes (probably CYP2D15) results in nonproportional increases in drug concentrations as the dose is increased up to 16 mg/kg orally; proportionality returns at 20 to 50 mg/kg orally. The injectable product remains potent for 28 days when prepared and stored according to labeled directions (amber vial at room temperature). Pain on injection may be common. Side effects delineated on the package insert include bone marrow hypoplasia in puppies younger than 11 (but not greater than 15) weeks of age.
Maropitant kinetics have been described in the cat after single subcutaneous and oral dosing and multiple subcutaneous dosing.22 The drug was well tolerated in cats (n = 6) during 15 days of subcutaneous administration at doses ranging from 0.5 to 5 mg/kg; one cat developed tremors at the 5 mg/kg dose. No changes occurred in clinical laboratory tests. Plasma maropitant concentrations increased proportionately with dose. After single intravenous dosing, the volume of distribution of maropitant in cats was 6.2 L/kg. Maximum drug concentration after oral and subcutaneous administration of 1 mg/kg in cats were 156 ng/mL and 269 ng/mL (with 50% coefficient of variabilility), respectively. The elimination half-life in cats was 13 to 17 hours; oral bioavailability varied from 50% to 117%. A dose of 1 mg/kg administered intravenously, orally, or subcutaneously prevented emesis induced by xylazine (0.44 mg/kg intramuscularly) and experimentally induced motion sickness.
Maropitant was approved for use in animals in Europe before the United States. It has proved effective for control of vomiting associated with drugs such as cisplatin, apomorphine, and morphine derivatives. Maropitant has been compared with metoclopramide (0.33 mg/kg every 8 hours subcutaneously in study one, 0.5 to 1 mg/kg/day in study two) in two multicenter, prospective, randomized, positively controlled clinical trials.30 Dogs (n = 64 in study one, 77 in study two) were at least 8 weeks of age and had been vomiting for at least 24 hours. Maropitant as studied at 1 mg/kg once daily subcutaneously in study one, and 0.5 to 1 mg/kg subcutaneously in study two with oral dosing of either maropitant or metoclopramide continued in study two until vomiting stopped or for up to 5 days. Vomiting caused by toxin ingestion or in patients with clinical signs indicating the need for acute surgical treatment were excluded. Causes of vomiting were multiple, including metabolic disorders, neoplasia, drug-induced reactions, food intolerance, and parvovirus. In both studies, maropitant was associated with a greater antiemetic response (discontinuation of vomiting) compared with metoclopramide.31
The comparative efficacy and safety of maropitant have been recently described for dogs on the basis of manufacturer-sponsored studies. It was compared (1 mg/kg) to placebo, metoclopramide (0.5 mg/kg subcutaneously), chlorpromazine (0.5 mg/kg subcutaneously), or ondansetron (0.5 mg/kg intravenously) in prevention of apomorphine-induced (0.1 mg/kg intravenously) vomiting. Efficacy in controlling vomiting either central or peripheral in origin was superior to that of chlorpromazine or metoclopramide but did not differ from ondansetron.32 Both safety and efficacy for prevention of emesis associated with motion sickness were assessed in dogs (n = 198) 16 weeks or older. Dogs received approximately 8 mg/kg orally. Data were collected from 26 different clinics in two different crossover randomized, placebo-controlled double-blinded trials with a 14-day washout period between trials. Vomiting was prevented in 86% or 77% of dogs dosed at 2 or 10 hours before a 60-minute car ride. No adverse events were described.
Vestibular Apparatus
Vomiting caused by motion sickness or inner ear disease is mediated by the vestibular apparatus (see Table 19-2). Motion sickness in dogs and cats can be controlled for several (8 to 12) hours by administration of antihistamines such as cyclizine hydrochloride, meclizine hydrochloride, or diphenhydramine hydrochloride (Figure 19-4; see also Figure 19-3). Effects may reflect, in part, sedative effects. In addition to direct effects on neural pathways arising in the vestibular apparatus, actions may also be independent of antihistaminic effects. These may include anticholinergic effects. Those drugs able to penetrate the blood–brain barrier may thus have effects at the vomiting center but generally only at higher doses. Drowsiness and xerostomia (dry mouth) are typical side effects that occur with use of this group of drugs in humans. Although phenothiazine antiemetics may be used to treat motion sickness (e.g., acepromazine), efficacy may reflect sedative rather than direct effects. Centrally active maropitant is approved for use in dogs to treat motion sickness (see preceding discussion) and has been used successfully in cats as well.
Drugs Active at the Chemoreceptor Trigger Zone
Phenothiazines
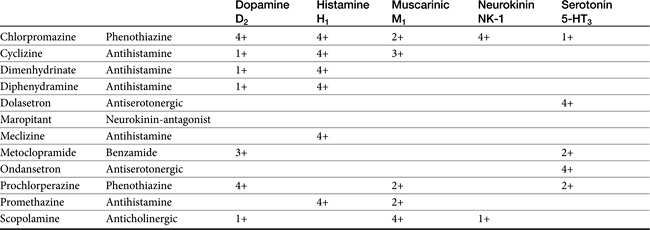
Phenothiazines are broad-spectrum antiemetics that control emesis induced by most central causes other than labyrinthine stimulation (see Figure 19-4). Their classification as broad reflects the variety of signals that serve as primary, secondary, and tertiary mediators. Phenothiazines block emesis mediated by the CTZ at low doses because of their antidopaminergic (D2) and, secondarily, antihistaminergic effects. Several phenothiazines are also characterized by weak antiserotinergic activity (see Table 19-2).24 At higher (perhaps nonpharmacologic) doses, their anticholinergic effects may also act at other central sites, including the vomiting center. A variety of phenothiazine derivatives (e.g., chlorpromazine, prochlorperazine, triflupromazine, perphenazine, trifluoperazine, and mepazine) are used in small animals as antiemetics. The primary adverse effects associated with their use as antiemetics are sedation (which contributes to their efficacy for motion sickness) and hypotension due to peripheral α-blockade. Selection of a particular phenothiazine may be based on avoidance of adverse reactions. Fluid replacement therapy should be instituted if necessary before use of a phenothiazine. The impact of phenothiazine derivatives on the seizure threshold and in epileptic dogs is discussed in more depth in see Chapter 27. In general, their use in epileptic animals may require caution but do not appear to be contraindicated.
Metoclopramide
Metoclopramide (see Figure 19-4) effectively blocks emesis mediated by the CTZ. Although its potent antagonism of dopamine was thought to be solely responsible for inhibition at the CTZ, metoclopramide is also a mixed 5-HT3 receptor antagonist/5-HT4 receptor agonist; emesis at high doses probably reflects 5-HT3 receptor antagonism.33 Metoclopramide effectively antagonizes apomorphine-induced emesis34 and is 20 times as potent as phenothiazines (although differences in efficacy have not been documented).35 The peripheral effects of metoclopramide on emesis resulting from prokinesis are discussed with the prokinetic drugs. Metoclopramide is indicated for control of emesis induced by a wide variety of blood-borne and peripheral causes.36,37 High doses of metoclopramide, particularly when combined with dexamethasone, have been used to treat emesis associated with cancer chemotherapy in human patients.38–40
Serotonin antagonists
Serotonin antagonists are useful for their antiemetic effects mediated at the CTZ, particularly those induced by chemotherapeutic agents (see Table 19-2).41 Unlike most other antiemetic drugs, antiserotonergics have no effects at other receptors, thus increasing the safety of those drugs selective for serotonin receptors. Ondansetron is a potent antiemetic and affects human cancer patients undergoing chemotherapy.23,33,42 It has also been used in small animals suffering from refractory vomiting that have not responded to other antiemetics. The efficacy of ondansetron reflects, in part, its active metabolite dolasetron, which is also available as an orally administered as well as an intravenous product. Dolasetron also is metabolized (reduced) to a metabolite characterized by greater activity for 5-HT3receptors compared with dolasetron.43 The pharmacokinetics have been reported for dolasetron and its reduced metabolite in dogs (n = 3).43 After intravenous administration of 2 mg/kg, clearance of the parent compound was 109 ± 41 mL/min/kg and volume of distribution was 0.83 ± 0.23 L/kg. After an oral dose of 5 mg/kg, Cmax of the parent compound was 219 ± 149 ng/mL at 0.17 hours. The elimination half-life was 0.15 ± 0.11 hour. The active metabolite appears to potentially double the AUC of active compound. Although oral bioavailability of the parent compound is less than 10%, it is probable that first-pass metabolism results in formation of the active, reduced metabolite: oral administration of 5 mg/kg radioactive dolasetron results in a Cmax of 700 ng/mL radioactivity (i.e., a combination of both radioactive parent and metabolite). Example uses of ondasetron include presurgical preparation, chemotherapy, and treatment of parvovirus infection; vomiting induced by hepatic lipidosis or GI irritation is less likely to respond. Ogilvie44 has reviewed the use of dolasetron in dogs.
Miscellaneous Antiemetics
Sedatives such as the barbiturates (phenobarbital) and the benzodiazepines have been used to control psychogenic and behavioral vomiting. Glucocorticoids and in particular dexamethasone are characterized by antiemetic effects, although the antiemetic mechanism of action is not understood.24 An antiinflammatory mechanism has been proposed. Glucocorticoids also appear to act in an additive or synergistic fashion when combined with other antiemetics. Both dexamethasone and methylprednisolone have been used in human patients to control vomiting associated with chemotherapy.
Peripherally Acting Antiemetics
Anticholinergics
Anticholinergic drugs that block muscarinic receptors in the emetic center also inhibit peripheral cholinergic transmission. Those anticholinergic drugs that do not cross the blood–brain barrier well are essentially peripheral in action and include glycopyrrolate, propantheline, isopropamide, and methscopolamine (which should not be used for cats). The ability of anticholinergics to suppress emesis is probably related to inhibition of afferent vagal impulses, relief of GI smooth muscle spasms, and inhibition of gastroenteric secretions. Delayed gastric emptying caused by these drugs may itself cause emesis, and anticholinergics should not be used for more than 3 days by the vomiting patient. Because of their anticholinergic properties, these drugs should not be used in combination with drugs whose actions depend on cholinergic activity in ganglion or smooth muscle. These include metoclopramide, cisapride, and the opioids.
Antiulcer Drugs
Pathophysiology of Gastrointestinal Ulceration
Gastroduodenal Ulceration
The events leading to gastroduodenal ulceration are complex and reflect interactions between acid-secreting and defense mechanisms of the GI mucosa.45,46 Regardless of the cause of GI erosion or ulceration, the basic pathologic mechanism is similar. Gastric acid secretion is a prerequisite for damage to the GI mucosa46,47 with luminal damage not occurring unless luminal pH is less than 7. Pepsin and bile acids can contribute to mucosal damage. Even though these chemicals are inherently caustic, mucosal damage generally does not occur in the face of normal mucosal cytoprotective mechanisms. These include but are not limited to secretion of bicarbonate and mucus and rapid epithelial turnover. Deceased mucosal blood flow can have a profound effect on the ability of the injured mucosa to heal itself. Drugs used to control or treat GI erosion and ulceration include those that inhibit gastric acid secretion or provide or facilitate other cytoprotective effects. The role of Helicobacter sp. in the pathogenesis of gastroduodenal ulceration in human patients has been well established, but its role in disease in animals is less well documented (see later discussion; e.g., IBDs).
Physiology of Gastric Acid Secretion
Gastric acid secretion occurs in four phases. The first three phases—referred to as cephalic, gastric, and intestinal—are stimulated by food and mediated by gastrin, which is the most potent secretagogue.48 Secretion is persistent during these phases, and gastric pH progressively decreases as nutrients traverse the GI tract. Gastrin secretion is inhibited as gastric pH declines to 3.5 and is completely inhibited at a pH of 1.5, to begin again only when pH approximates 3 to 3.5. The fourth phase of gastric acid secretion is basal and occurs in the absence of external stimuli. The amount of basal secretion varies among animals. In humans basal secretion follows a circadian rhythm, reaching a peak at midnight and a nadir at 7 am.49 As a model for the study of antisecretory drugs, information can be found regarding basal and responsive gastric acid secretion in dogs.50,51
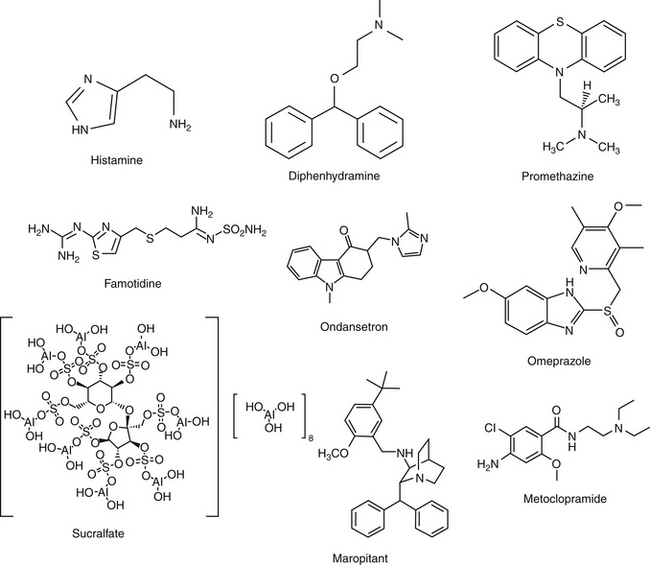
Gastric acid secretion at the cellular level involves the generation and subsequent secretion of hydrogen ions by the parietal (oxyntic) cells of the gastric mucosa. Responses are controlled through chemical signals interacting with corresponding receptors located on the basolateral membrane of parietal cells. Central and peripheral signals stimulating gastric acid secretion include endocrine (gastrin: CCK), paracrine (histamine: H2), and neuronal (ACh: M3) (Figure 19-5).49 In addition to its direct effects, acetycholine indirectly increases release of gastrin from G cells and histamine from enterochromaffin cells. Of the mediators increasing gastric acid secretion, gastrin is the most potent, although its effects are mediated indirectly through stimulation of histamine receptors, particularly on enterochromaffin cells.48 Somatostatin, which inhibits gastric acid secretion, is released from D cells when gastric pH is less than 3. In humans the effects of Helicobacter spp. may reflect, in part, the ability of these organisms to decrease D cells. The hydrogen ion pump, located at the apical membrane and associated with the smooth endoplasmic reticulum, is unique in that it is a hydrogen–potassium ATPase exchange system. Three distinct pathways are capable of stimulating gastric acid. Each acts through chemical mediators that in turn interact with receptors on the parietal cell membrane.49 H2 receptors are linked to adenylyl cyclase and cyclic AMP.48 Of these, the ACh pathway appears to be less important in small animals.
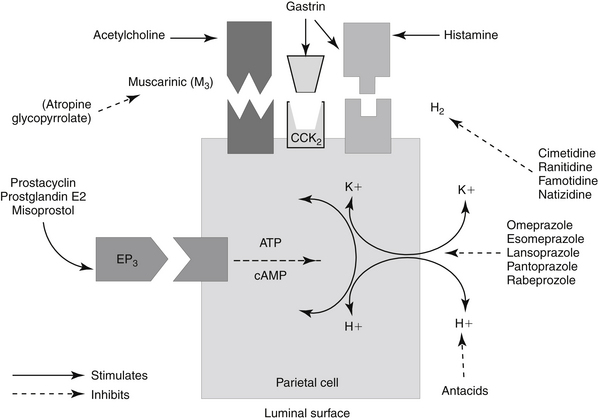
Intracellular messengers mediating gastric acid secretion vary with the receptor stimulated. Histamine increases cAMP production, which subsequently activates the adenylyl cyclase cAMP-dependent protein kinases. Gastrin and muscarinic stimulation by cholinergic drugs increased cytosolic calcium, through inositol phosphate pathways. Both pathways activate an H+, K+-ATPase proton pump that exchanges hydrogen and potassium across the parietal cell membrane. Prostaglandins of the E series serve to modulate these effects, inhibiting gastric acid secretion by blocking cAMP production through EP3 receptors, also on parietal cells.48,49 The impact of opioid receptors on gastric acid secretion is discussed with drugs targeting intestinal secretion.
Mucosal Defenses
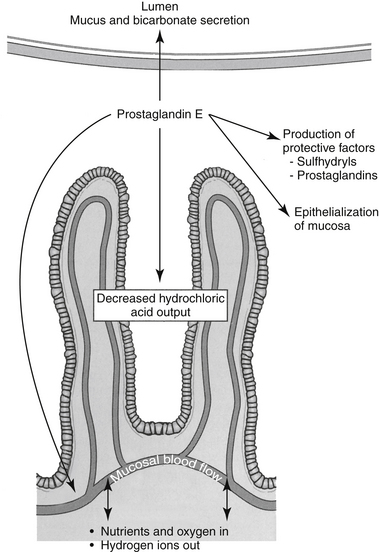
The primary mucosal defense of the esophagus reflects increased lower esophageal sphincter tone. Defenses of the GI mucosa require sufficient mucosal blood flow and act to prevent or repair GI ulceration (Figure 19-6).52–54 These include (1) secretion of bicarbonate into the lumen and neutralization of hydrochloric acid in the lumen; (2) secretion of a thick, insoluble, alkaline mucus that traps and neutralizes inward-moving hydrogen ions and protects against macromolecules such as pepsin; (3) a gastric epithelial barrier composed of active phospholipids, a lipoprotein cell membrane, and tight junctional complexes, all of which prevent hydrogen ion back diffusion; (4) mucosal blood flow, which first provides nutrients and oxygen to mucosal cells and second removes hydrogen ions that have penetrated the gastric barrier; (5) rapid replication of mucosal epithelial cells; and (6) production of cytoprotective agents. Many of these effects reflect local secretion of prostaglandin E2 and I2, important defense mechanisms. They modulate hydrochloric acid secretion, increase bicarbonate and mucus production, and enhance mucosal blood flow and epithelialization.55,56 Sulfhydryls also produced locally may act as scavengers of oxygen and other tissue-damaging radicals.57
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


